Case History
The patient is a 77 year old man with a longstanding history of increased white blood cell (WBC) count who presented with a new rash and increasing absolute lymphocytosis.
Labs

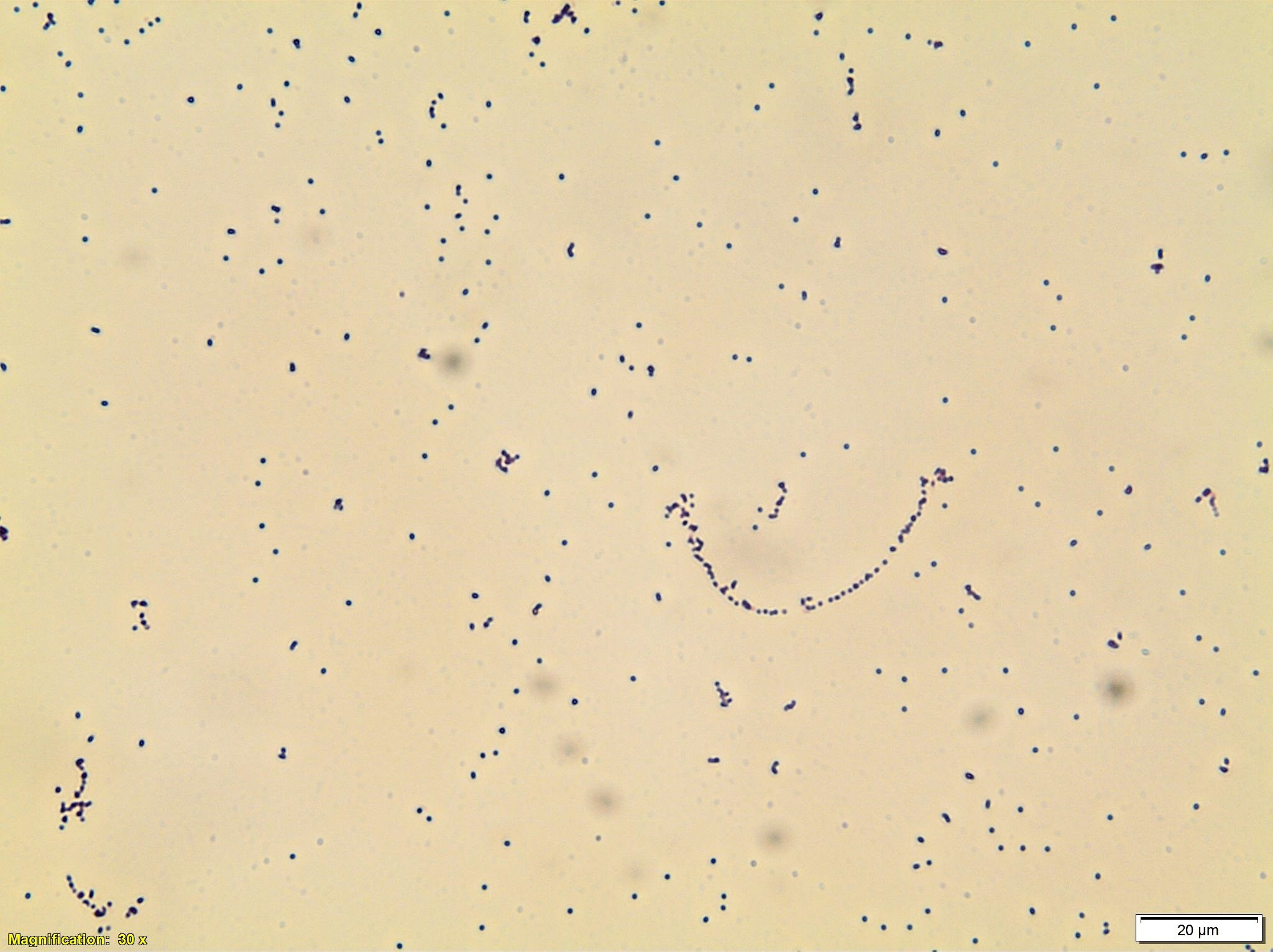
Peripheral Blood Smear

Peripheral blood smear shows small to medium-sized lymphocytes with basophilic cytoplasm, cytoplasmic protrusions or blebs, round to oval nuclei with indented nuclear contours and some cells with prominent nucleoli.
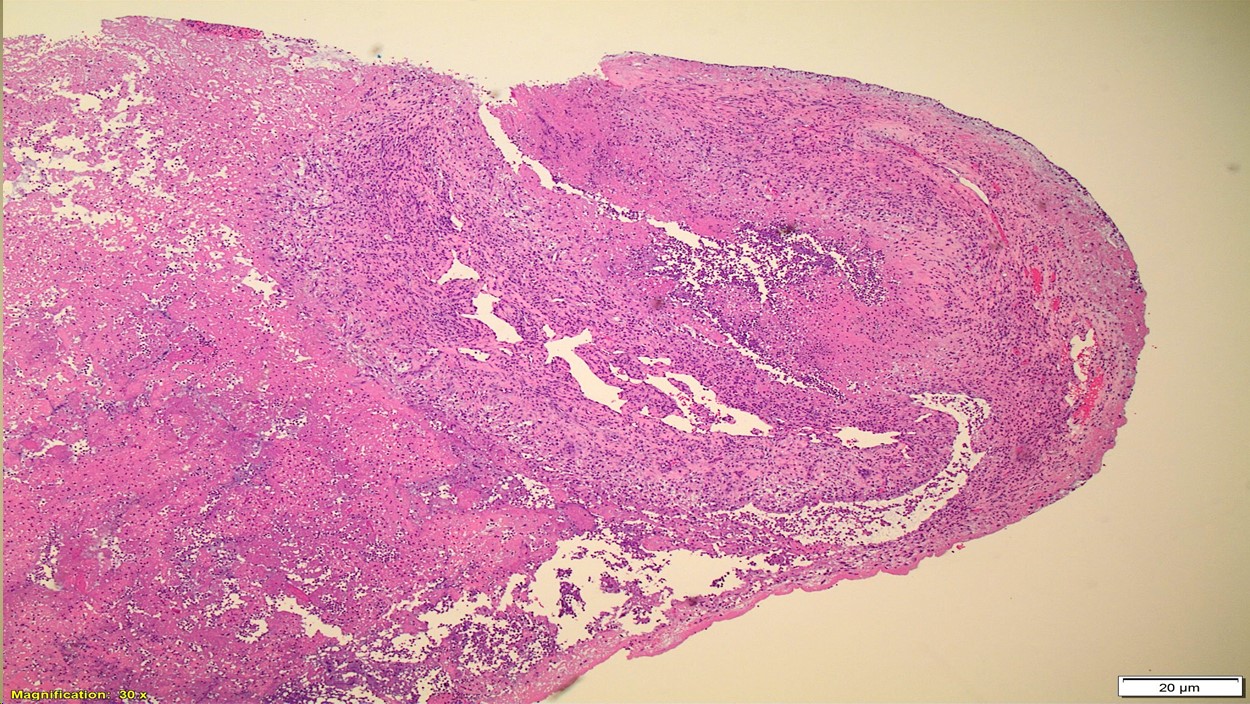
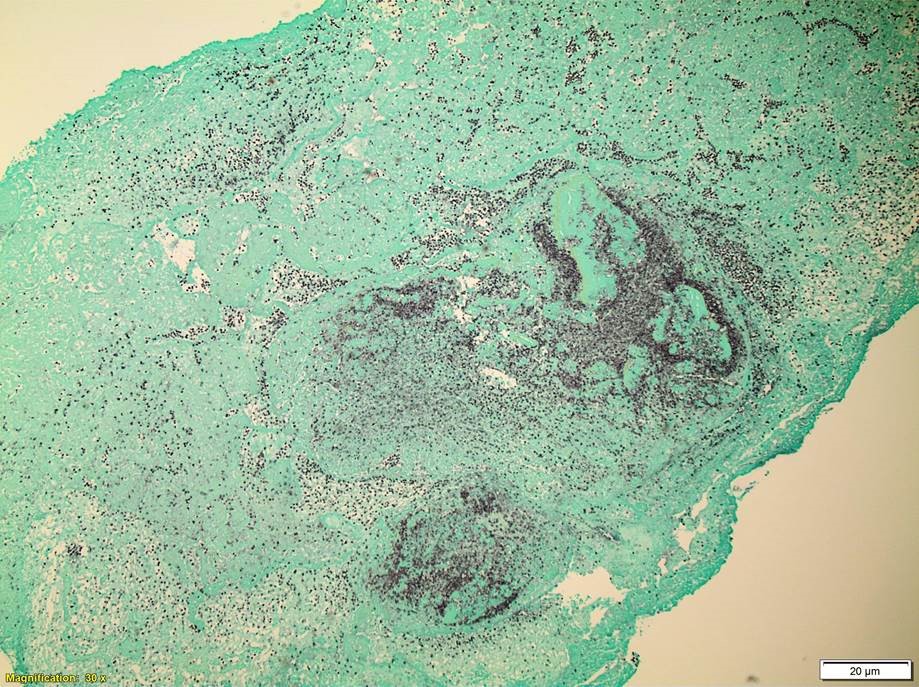
Bone Marrow Biopsy

Bone marrow aspirate (top left) shows increased lymphocytes with similar features to those seen in the peripheral blood. The core biopsy (top right) shows an abnormal lymphocytic infiltrate. By immunohistochemistry, CD3 highlights markedly increased interstitial T-lymphocytes (30-40%) that predominantly express CD4. CD8 highlights only few scattered T-cells.
Flow Cytometry

Concurrent flow cytometry identifies an expanded population of lymphocytes comprising 73% of the total cellularity. Of the lymphocytes, 98% are T-cells. The T-cell population is almost entirely composed of CD4 positive cells (CD4/8 ratio = 301). The T-cells show expression of TCR (a/b), normal T-cell antigens CD3, CD2, CD5 and CD7 and express CD52 (bright).
Cytogenetics

Concurrent chromosome analysis shows that 90% of the metaphase bone marrow cells examined have a complex abnormal karyotype with a paracentric inversion of chromosome 14 that results in the TRA/D/TCL1 gene rearrangement. There is also a rearrangement resulting in three copies of 8q with partial loss of 8p as well as other chromosome aberrations.
Diagnosis
Altogether, the presence of an abnormal CD4 positive and CD52 (bright) lymphocyte population with the characteristic cytogenetic finding of inv(14), is diagnostic of T-cell prolymphocytic leukemia (T-PLL). This patient’s course is unusual in that he initially presented with indolent disease that ultimately progressed. The lymphocyte morphology was also somewhat atypical in that only occasional cells had prominent nucleoli. This is consistent with the “small cell variant” of T-PLL.
Discussion
T-PLL is generally an aggressive disorder characterized by small to medium sized mature T-cells that are found in the peripheral blood, bone marrow, lymph nodes, spleen, liver and sometimes skin. T-PLL is rare and occurs in adults usually over 30 years old. The clinical presentation includes a lymphocytosis, often >100 x 10^9/L, hepatosplenomegaly and lymphadenopathy. Serous effusions and skin infiltration can be seen in a subset of cases. On microscopy, the cells are usually small to medium in size with basophilic cytoplasm, round to irregular nuclei and visible nucleoli. Characteristic cytoplasmic blebs or protrusions are a common feature. The immunophenotype is of a mature T-cell and cells are positive for CD2, CD3, CD5 and CD7. They are negative for TdT and CD1a. Another characteristic feature is bright expression of CD52. Sixty percent of cases are positive for CD4, while 25% show double expression of CD4 and CD8. The most frequent chromosome abnormality is inversion of chromosome 14 at q11 and q32, which is seen in 80% of patients. Translocations involving chromosome X and 14 are also seen, as well as abnormalities of chromosome 8. The overall prognosis is generally poor with a median survival of 1-2 years. Patients with expression of CD52 may respond well to the monoclonal anti-CD52 antibody alemtuzumab, but other treatment options are limited.1
The small cell variant (SV) of T-cell prolymphocytic leukemia was once referred to as T-cell chronic lymphocytic leukemia due to a predominant population of small lymphocytes with condensed chromatin and lack of conspicuous nucleoli. In addition, unlike the aggressive course seen in most patients with T-PLL, patients with this morphology tended to have an indolent or more chronic disease course. Eventually, it became clear that this was merely a variant of T-PLL due to similar immunophenotypic and cytogenetic findings. Ultimately, the term T-cell CLL was retired from use.2
In a comparison of patients with SV T-PLL to three large studies of classic T-PLL patients, the SV patients were found to have a higher frequency of a normal karyotype and increased double negative (CD4-/CD8) immunophenotype. Interestingly, 38% of the SV patients did not receive treatment for the entire duration of follow-up, while 19% required treatment after initially just being observed. This time period ranged between 2 months to 3 years. The remaining patients were treated at diagnosis. Most of the patients ultimately progressed and the cause of death was disease progression in 86% of the patients who died during follow-up. Overall, SV T-PLL tended to show less aggressive clinical behavior than classic T-PLL, however many aggressive cases of patients with the small cell variant have been seen. Likewise, more indolent cases of classic T-PLL featuring cells with larger nuclei with prominent nucleoli have also been described.2
While cases of SV T-PLL may initially present with more indolent disease, they almost always progress to a similarly aggressive disease course as seen in classic T-PLL. T-PLL is generally resistant to most conventional chemotherapies. As mentioned earlier, cases of T-PLL tend to express bright CD52, which is a glycoprotein present on the surface of mature lymphocytes. CAMPATH-1H is an anti-CD52 monoclonal antibody that may result in complement-mediated lysis and antibody-dependent cellular cytotoxicity. In a study by Dearden et. al., thirty-nine patients with T-PLL received CAMPATH-1H treatments. The overall response rate was 76% with 60% achieving complete remission. These rates are significantly higher than those reported for conventional therapies like CHOP. Unfortunately, almost all of the patients ultimately progressed and all but 2 had relapsed following 1 year of therapy. This indicates that CAMPATH-1H is good for first line therapy, but is not a curative treatment for this aggressive and most often deadly disease. 3
References
- Swerdlow SH, Campo E, Harris NL, et al. WHO Classification of Tumours of Haematopoetic and Lymphoid Tissues (Revised 4th edition). IARC: Lyon 2017.
- A. Rashidi and S. Fisher. T-cell chronic lymphocytic leukemia or small-cell variant of T-cell prolymphocytic leukemia: a historical perspective and search for consensus. European Journal of Haematology. 2015(Vol 95).
- C. Dearden, E. Matutes and B. Cazin, et. al. High remission rate in T-cell prolymphocytic leukemia with CAMPATH-1H. Blood. 2001(98)1721-1726.

–Chelsea Marcus, MD is a Hematopathology Fellow at Beth Israel Deaconess Medical Center in Boston, MA. She has a particular interest in High-grade B-Cell lymphomas and the genetic alterations of these lymphomas.