Inspections are a great learning opportunity. Our recent American Association of Blood and Biotherapies (AABB) introduced us to the risk analysis requirement in its 2024 checklist. Though the requirement is specifically stated in 3.7 Information Systems #13, the requirement can be applied to instruments as well as software. The update is significant as it emphasizes the need to proactively identify, assess, and mitigate risks associated with introducing or modifying software and instruments within the laboratory setting.
AABB-accredited laboratories will need to review their validation procedures to ensure they include a risk analysis with mitigation for identified risks. There are a few steps to conducting a risk analysis.
First, clearly define the changes being made. This could include new software installations, updates to existing software, introduction of new instruments, or modifications to current equipment. Understanding the full scope of changes is essential for a comprehensive risk analysis.
Secondly, if possible, form a multidisciplinary team that includes IT specialists, laboratory managers, quality assurance personnel, and end-users. In reality, for many small to medium laboratories, frequently one person fills more than one role. It’s conceivable for the laboratory manager to be the quality person and an end user since they often may have to fill in during staff shortages.
Next, identify the risks. Structured techniques like Failure Modes and Effects Analysis (FMEA) can be used to identify potential risks. Consider software-based risks such as data loss or corruption, system incompatibility, user interface issues, and cybersecurity vulnerabilities. Calibration errors, operational failures, compatibility with existing systems, and maintenance requirements are some instrument-based risks that should be considered. And, of course, human-based risks involving user errors, poor or insufficient training, and workflow disruptions.
The fourth step would be to assess each identified risk, assessing its potential impact on laboratory operations and patient safety and evaluating the likelihood of occurrence and the severity of consequences if the risk materializes. Use a risk matrix to categorize risks as low, medium, or high.
For each high or medium risk identified, develop mitigation strategies to reduce the likelihood of occurrence or minimize the impact. Some examples are increasing training programs, additional testing, or developing contingency plans for failures.
Ensure the entire risk analysis process is documented, including the identified risks, evaluation results, and mitigation strategies. Documentation is crucial for compliance with the AABB checklist and is a reference for future audits or inspections.
The risk analysis requirement in the AABB’s 2024 checklist underscores the importance of proactive risk management in clinical laboratories. Through the implementation of the outlined steps, laboratories can not only meet this requirement but also enhance their operational resilience and commitment to patient safety. Conducting a thorough risk analysis for software and instrument changes is an investment in the quality and reliability of laboratory services, ultimately contributing to better patient outcomes.
-Darryl Elzie is the Regulatory Affairs Manager Inova Blood Donor Services. He has been an ASCP Medical Laboratory Scientist for over 25 years, performing CAP inspections for two decades. He has held the roles of laboratory generalist, chemistry senior technologist, and quality consultant. He has a Master’s in Healthcare Administration from Ashford University, a Doctorate of Psychology from The University of the Rockies, and is a Certified Quality Auditor (ASQ). Inova Blood Donor Services is the largest hospital-based blood center in the nation. Dr. Elzie is also a Counselor and Life Coach at issueslifecoaching.com.
On May 11, 2023, the FDA’s Center for Biologics Evaluation and Research (CBER) issued guidance updating the blood donor history questionnaire (DHQ). Also known as the DHQ v4.0. The hope was that creating a gender-neutral questionnaire would increase the number of people eligible to donate, improving the nation’s blood supply.
After 3 months, has it made a difference?
The DHQ v4.0 is a series of questions asking potential donors about their lifestyle activities and travel to assess whether they are eligible to donate blood. The questionnaire is a risk-based model and is a critical step in ensuring the safety and potency of the nation’s blood supply.
As knowledge and understanding about disease increases along with the ongoing need to maintain an adequate blood supply, the FDA, in conjunction with the Association for the Advancement of Blood and Biotherapies (AABB), reviewed the restrictions or limitations on groups or individuals who may be allowed to donate with the goal of increasing the pool of eligible donors.
The DHQ v3.0 contains gender-specific questions impacting the eligibility of LGBTQ members. The new DHQ v4.0 was developed to be gender-neutral. There was concern that the new questions (especially the follow-up if any were answered yes) would be uncomfortable, but they were necessary to assess every potential donor the same.
“Some of the follow-up questions can seem to be a bit personal,” states Marvin Opulencia, Donor Operation Trainer at Inova Blood Donor Services (IBDS).
But Marvin thinks the change was a good thing because the questionnaire is no longer gender-specific and makes the process easier. “Some of my friends are members of the LGBTQ community, and now they are able to donate. I’m happy about that.”
Recognizing the difficulty and sensitivity inherent in the impacted topics, the FDA did not issue a deadline for implementation but has allowed blood donation centers to integrate the new guidance at their own pace. However, there were some blood donor centers that were ready to move forward with the recommendations. Their experience tells us that it is still too early to evaluate the effect of the change.
Nicholas Lilly, Interim Director of IBDS in Northern Virginia, believes we still don’t know the overall impact of the DHQ v4.0. “Inova Health System welcomes and supports initiatives improving the diversity, inclusiveness, and equity of our services. That’s why we were one of the first to implement the DHQ v4.0 in June. We serve the DMV area (D.C., Maryland, and Virginia), which is a highly diverse community, and so we saw the new guidance as a continuation of our vision and goals.”
With three months of data to evaluate, Director Lilly still doesn’t know what impact the change has on the blood supply. “Though we have received no negative feedback from our clients, we still haven’t seen a net increase in the number of donors. We have seen more non-binary people donate, but overall, there has not been a noticeable increase in donations. But it’s still early.”
It was expected that changing the Individual Donor Assessment to a more gender-neutral questionnaire would generate a bit of consternation and questions regarding whether it was in the nation’s best interest. However, so far, it was just one more step toward allowing everyone to make a difference.
Stated simply, Blood Saves Lives and is desperately needed. Now everyone has the opportunity to donate. Limitations or restrictions should be determined by science-backed non-judgmental research. That’s what the DHQ v4.0 is… that’s the difference it makes.
–Darryl Elzie is a Quality Consultant for Inova Blood Donor Services. He has been an ASCP Medical Technologist for over 25 years, performing CAP inspections for 15+ years. He has held the roles of laboratory generalist and chemistry senior technologist. He has a Master’s in Healthcare Administration from Ashford University, a Doctorate of Psychology from The University of the Rockies, and is a Certified Quality Auditor (ASQ). Inova Blood Donor Services is the largest hospital-based blood center in the nation. Dr. Elzie is also a Counselor and Life Coach at issueslifecoaching.com.
One of my favorite things about working in Hematology is handling those “difficult” samples. You know the ones. The one that some techs put aside to work on “later,” or they might decide it’s time to take a break when they see them coming. I love investigating and working on these interesting but perhaps uncooperative samples. At times this involves running samples in different modes, making new slides or albumin smears, and diluting samples. At other times, we investigate a delta or unusual results by checking patient diagnosis and previous results or by calling the care provider for more information and clues to help us resolve the problem.
I’m sure you’ve all seen the sayings “Without the Lab, you’re only guessing” and “Laboratory Professionals get results.” Physicians rely on the lab every day for information used to help diagnose and treat patients. Therefore, our goal is to deliver to the care provider the best possible results in a timely manner. Which means that we don’t just report results because that’s the answer the instrument gave us. With today’s instruments and middleware, we get very accurate and precise results, and about 85% or more of hematology specimens autovalidate. This is important because it leaves us time to work on those specimens with flags, and discrepancies; the ones that need a little more time and attention.
When faced with unusual or conflicting results, we first need to ask ourselves if we are dealing with a spurious sample, interfering substances or true abnormal results. Many labs today use middleware that will give the operator alerts when a sample needs to be investigated. These alerts give us suggestions as to how to handle the specimen but are usually short phrases triggered by certain values or flags and cannot be all encompassing. Operator alerts cannot tell us all the steps we may need to follow to resolve, for example, deltas, platelet clumps, abnormal scattergrams or a possible cold agglutinin. The alerts are great guidelines but it is often necessary to do more. We may need to refer to procedure manuals for SOPS or check instrument manuals or technical bulletins to decide how to handle these specimens. Sometimes we need to be detectives to report the most accurate results. We must review results with a critical eye, use all that “stuff” we learned in school, and be able to make educated decisions based on this investigation.
In my experience, one of the most common troublesome and perhaps misunderstood specimens I see is the one with a “hemoglobin (Hgb) interference” flag. An instrument flag “suspect, turbidity /Hgb interference?” is generally initiated when the MCHC is above a certain value. In our hematology lab, we see this flag when the MCHC is above 37.5 g/dL. What this is telling us is that turbidity may be present in the diluted and lysed sample. This turbidity can interfere with the Hgb detection light path and falsely increase the Hgb. Because the MCH and MCHC are calculated using the Hgb, these parameters are also affected. BUT, an MCHC >37.5 g/dL is not always something that can be or that needs to be corrected. With any parameter 95% of normal values will fall within 2SD of the mean. This means that 5% of normal healthy individuals have MCHC results <32 g/dL or >36 g/dL, and a few may have an MCHC over 37.5 g/dL. An MCHC >37.5 g/dL therefore can indicate a normal specimen, such as in a healthy young male with a Hgb at the high end of the reference range. High MCHCs can also be seen routinely in specimens from patients with spherocytosis or hemoglobinopathies such as Hgb SS, Hgb SC or Hgb C disease. In these conditions the RBCs are hyperdense due to altered surface volume and this leads to a high MCHC.
On our instrument, an MCHC >37.5 g/dL will cause a Hgb/Turbidity flag. An asterisk (*) will appear next to the Hgb, MCH and MCHC. The middleware triggers an operator alert that says “MCHC >37.5. Incubate at 37C for 30 mins. Evaluate for lipemia, icterus, hemolysis, Plasma replacement if indicated, rerun”. So, what’s the first thing to do?? Incubate? Hold on…not so fast. This is one of those instances where hematology is not just black and white. This operator alert is giving us suggestions of how to handle a specimen, but techs need to evaluate the specimen before jumping on the ‘cold’ wagon. Incubating will usually help resolve a cold agglutinin, but won’t help with a sickle cell specimen, or resolve one that’s icteric or lipemic. A grossly hemolyzed sample can give a spurious high MCHC result and, if so, needs to be recollected, not warmed. Putting a specimen that’s hemolyzed or lipemic or icteric in the heating block for 30 or more minutes would only delay reporting of results. My first case example involves a 45 year old female. The MCHC on initial run was 38.1 and the specimen gave a Hgb turbidity flag. The sample was incubated and rerun several times. After 1 hour of incubation, the MCHC was reported as 37.1 with a comment “repeated after warming for 1 hour at 37C”. In this case the patient was a known sickle cell patient. Previous results show that this patient’s MCHC is typically high and previously reported results ranged from 36.1- 37.8 g/dL. When evaluating a specimen with a high MCHC it is important to check the pattern of results. In this case the MCHC was high but the MCV was low. This does not fit the pattern for a cold agglutinin. As noted above, super dense RBCs in sickle cell patients may cause a high MCHC. This specimen was warmed, and even though the MCHC was a bit lower after warming, it would have been acceptable to report the original run MCHC. Checking patient history and previous results, and reviewing the smear for morphology would have allowed these results to be reported in a timely fashion. The operator alert does say “incubate the specimen” but it also says to evaluate. Be sure to check the MCV and MCHC along with patient history before warming specimens that don’t fit the pattern of a cold agglutinin.
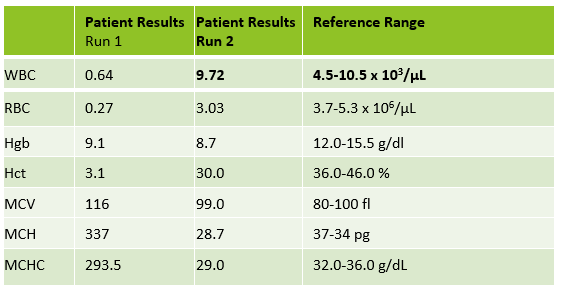
Table 1. Case 1 CBC. The patient is a 45 year old known sickle cell patient.
The second example is from a 75 year old male. The CBC flagged Hgb turbidity with an MCHC of 45.8 g/dL. The MCHC >37.5 operator alert triggered Checking the pattern of results for the indicies, the MCHC was very high and the MCV was low. In a specimen with a low or normal MCV and a high MCHC, lipemia, icterus, abnormal proteins or severe leukocytosis can be affecting the Hgb. On evaluation, this sample’s Hgb and Hct did not meet the ‘rule of 3’. The rules of 3 are now generally recognized to be valid only for samples when the RBCs are normal, but the * here is telling us that there is an interference affecting the Hgb. In these cases it is valuable to know what the interference is so we know how to handle the specimen. By spinning down a small aliquot, (or asking chemistry!) we can investigate for lipemia or icterus. The specimen was found to be grossly lipemic. Flagging guidelines for lipemic specimens suggest diluting the specimen 1:5 and rerunning. Alternately, with severely lipemic or icteric samples, plasma replacement procedure may be necessary to correct the results. In this case, a plasma replacement was performed. After a plasma replacement, the WBC, RBC, Hct, MCV and platelet count are reported from the original run. The Hgb interference is what was causing the problem. Thus, when you correct the Hgb you must always correct any indicies that are calculated with the Hgb. The Hgb from the plasma replacement sample is used and the MCH and MCHC are recalculated. Notice that the new lower Hgb value now matches the Hct.
Table 2. Case 2, a 75 year old male with lipemic specimen. Plasma replacement performed. WBC, RBC, Hct, MCV, and Plt were reported from original run. Hgb was reported from plasma replacement sample. MCH and MCHC were recalculated.
Case 3 is a sample from an 80 year old woman. This was an interesting sample because there were multiple things going on here. This patient had an initial result with a high MCHC and MCH, with decreased RBC and Hct. In this patient the initial WBC was 0.64 and the RBC was 0.31. The Hgb of 9.1 /dL was less than the Hct of 3.1 %. MCV was 116 fl and the MCHC was 293.5 g/dL! In specimens with a high MCV and high MCHC we can suspect a cold agglutinin. When the MCV is very high it is because the RBCs are going through the aperture as one big bunch and this is measured as the size of one RBC. Often the Hct is less than the Hgb. Sometimes the RBC and Hct are so low that it causes the MCV to be appear within normal range. On our instrument, a RBC count of <0.5 x106/μL will give a flag “abnormal RBC scattergram” but no other indicies related flags are generated, so we didn’t even get an operator alert to evaluate the MCHC. But, it’s clear there is something very wrong with these results. Warming the sample is used to loosen clumping of RBCs, which lowers the MCV and allows the RBCs to be counted. Make a smear to examine for RBC clumping and look at the sample tube. Many cold agglutinin samples will appear to be ‘grainy’ or have agglutination along the side of the tube. This is the time when we want to incubate the sample. To resolve a cold agglutinin, warming the sample is necessary. Sometime 30 minutes is enough, sometime they need to be incubated longer. Some cold agglutinins are so strong that after incubation a dilution or plasma replacement still needs to be done. Warming this sample did not lower the MCHC. After incubating, I diluted this sample, and also did a plasma replacement to see how results would compare. The new results matched. This sample took a bit more time than others but the cold agglutinin was resolved and we were able to report valid results.
Table 3. CBC results from 80 year old woman with cold agglutinin. Image 1. Tube from cold agglutinin specimen. Note agglutination in sample along sides of tube.
There are other factors that can affect the Hct or Hgb and cause a high MCHC. Icteric specimens act much like lipemic ones and the Hgb can be corrected with dilution or a plasma replacement. An electrolyte balance can affect the Hct. Abnormal proteins and severe leukocytosis can affect the Hgb. Grossly hemolyzed samples can have a high MCHC. It is important to evaluate the indicies in these samples and correlate the values with previous results and patient history. What concerns me is that I have seen samples being warmed that do not match the indicies patterns for cold agglutinins. I have seen samples from sickle cell patients signed out with a comment “warmed at 37C. Possible cold agglutinin.” I have seen lipemic or icteric samples that are reported out with high MCHCs, erroneously high Hgb or parameters that are not reported at all. While warming these samples may actually lower the MCHC a bit, it still usually remains on the high side and does not give us the clean results that dilution or plasma replacement will. A little extra time looking at the indicies can give us important clues as to how to handle these samples. Doctors use our results every day to make patient care decisions. We need to make sure that we are making decisions every day to give them the best possible results so that patients can get the best care possible.
Table 4. Evaluating high MCHC specimens.
References
Costa, B. M. B., Vellés, M. C., Viana, M. M. F. B., & Rebelo, C. I. M. (2018). Interference of cold agglutinin autoantibodies in erythrogram interpretation: a case report and literature review. Jornal Brasileiro De Patologia e MedicinaLaboratorial, 54(4). doi: 10.5935/1676-2444.20180043
Sysmex USA. XN-Series Flagging Interpretation Guide. Document Number: 1166-LSS, Rev. 6, March 2021
It’s not Black and White: Unraveling the puzzles of Hematology. Becky Socha MS, BB, MLS(ASCP) Mercy Medical Center, Baltimore, MD
-Becky Socha, MS, MLS(ASCP)CMBBCM graduated from Merrimack College in N. Andover, Massachusetts with a BS in Medical Technology and completed her MS in Clinical Laboratory Sciences at the University of Massachusetts, Lowell. She has worked as a Medical Technologist for over 40 years and has taught as an adjunct faculty member at Merrimack College, UMass Lowell and Stevenson University for over 20 years. She has worked in all areas of the clinical laboratory, but has a special interest in Hematology and Blood Banking. She currently works at Mercy Medical Center in Baltimore, Md. When she’s not busy being a mad scientist, she can be found outside riding her bicycle.
A newborn, healthy, full term, male child, was born with bruising on his left thigh and developed petechiae and purpuric hemorrhages several hours after birth. The baby was moved to the NICU for observation and a CBC was ordered by the NICU provider.
WBC, RBC, Hgb, Hct and indicies were normal
Platelet count 58 x103/μL
Baby exhibited no symptoms of sepsis
Smear reviewed with no platelet clumping observed
The mother is a 28 year old, gravida 1, para 1 with normal CBC and platelet count. Her prenatal history was unremarkable. She has no history of immune thrombocytopenia (ITP) and no history of being prescribed drugs known to be associated with drug induced thrombocytopenia
Thrombocytopenia is not an uncommon finding in neonates, particularly in the neonatal intensive care unit (NICU). In preterm infants, the most common causes of thrombocytopenia are complications of pregnancy, including pregnancy-induced hypertension (PIH), intrauterine growth retardation, preeclampsia ,and HELLP syndrome (hemolytic anemia, elevated liver enzymes, low platelet count). Examination of a peripheral smear in these patients will typically reveal neutropenia with densely packed red cells, increased nucleated RBCs and deceased platelet estimate. These placental insufficiency cases typically occur within the first 72 hours of life, platelet counts are >50 x 103/μL, resolve without treatment and require no further investigation. On the other hand, thrombocytopenia in preterm infants that develops after 72 hours is most likely due to sepsis or necrotizing enterocolitis and requires investigation and treatment.2
In an otherwise healthy appearing full term infant, the most common cause of thrombocytopenia in the first 72 hours of life is neonatal alloimmune thrombocytopenia (NAIT). When a platelet count drops below 150 x 103/L in these newborns, it is important to investigate the thrombocytopenia. The first step is to always check a peripheral smear for clumping to rule out spurious thrombocytopenia. With a low platelet count and the absence of spurious thrombocytopenia, NAIT can be suspected. This condition is similar in pathogenesis to hemolytic disease of the fetus and newborn (HDFN), and is caused by an incompatibility in human platelet antigens between mother and baby. In about 80% of cases, the mother is found to be HPA-1b and the father and baby are HPA-1a.1 The mother forms anti-HPA-1a which crosses the placenta and destroys the fetus’ platelets. Most cases of NAIT are asymptomatic, or cause only mild bleeding, and resolve in 1-2 weeks.1
Although many cases of NAIT are mild, it is important to recognize because it can be a life-threatening disorder. With more severe thrombocytopenia, in both premature and full term infants, NAIT can result in intracranial bleeding either before birth or shortly after birth. NAIT can also cause long term neurologic complications. Therefore, when a neonate is suspected to have NAIT, he should be screened for intracranial hemorrhage. Since mothers are most often found to have anti- HPA-1a, and the second most commonly found antibody is anti-HPA-5b, neonates with platelet counts <30 x 103/L should be transfused with antigen matched or HPA-1a and HPA-5b negative, CMV negative, single donor apheresis platelets.
It is important to note that NAIT can occur in a first pregnancy but subsequent pregnancies are usually more severely affected. In confirming NAIT after a first delivery or monitoring a subsequent pregnancy, serological testing should be done on both parents to determine the risk of having an infant born with NAIT. If the father is homozygous for the antigen which the mother lacks, 100% of infants would be at risk. If the father is heterozygous, an infant would have a 50% chance of inheriting the antigen from the father.
NAIT in a first pregnancy is typically unrecognized until after birth. Some groups have advocated for routine prenatal screening for NAIT in all pregnant women, but this is costly and still debated. It is agreed that after an affected first child, subsequent pregnancies should be monitored closely. In at risk pregnancies, weekly antenatal IVIg infusions should be used during pregnancy to help prevent fetal bleeding.3
The mother in this case was tested and found to be HPA-1a negative with anti-HPA-1a. The father was also tested and found to be HPA-1a positive. The infant’s platelet counts began to increase at 7 days, with no further bleeding. The mother was referred to a NAIT specialty team for future pregnancies.
Subarna Chakravorty and Irene Roberts. How I manage neonatal thrombocytopenia . Blackwell Publishing Ltd, British Journal of Haematology. 2011; 156, 155–162
-Becky Socha, MS, MLS(ASCP)CM BB CM graduated from Merrimack College in N. Andover, Massachusetts with a BS in Medical Technology and completed her MS in Clinical Laboratory Sciences at the University of Massachusetts, Lowell. She has worked as a Medical Technologist for over 30 years. She’s worked in all areas of the clinical laboratory, but has a special interest in Hematology and Blood Banking. When she’s not busy being a mad scientist, she can be found outside riding her bicycle.
Hello again! The last case study was an example of a patient with a loss of allele at two STR loci on a shared chromosome. Today, I wanted to share an interesting and unusual case that we monitor in our lab. This case explores the use of cord bloods as the source of the donor, and in this case, a double cord blood transplant.
Cord blood (CB) unit transplants can be advantageous over other donor sources, such as bone marrow or peripheral blood. The Leukemia and Lymphoma Society summarizes these advantages well, with some being their availability (CB can be prescreened/tested and then frozen for use when needed – decreasing the risk of disease transmission), less-strict HLA matching requirements, decreased graft versus host disease (GVHD) occurrence and severity, long-term storage (CB over 10 years old has been successfully transplanted), increased diversity of donors, and reduced risk of disease relapse, to name a few.2, 3
CB also has its disadvantages, some include: less stem cells for engraftment which leads to longer engraftment times, these longer engraftment times lead to longer immunological recovery and a higher risk of infection, less available clinical data relative to stem cell and bone marrow transplants (newer procedure comparatively in transplant), and no additional cells for infusions later on in treatment. Further, selecting the best cords for transplant can be challenging due to the static variables of a CB (again, there is no donor to go back and get more cells). Considering all that CB has to offer, haplo-identical transplants are preferred in the U.S. over CB transplants. 2,3,4
Before the University of Minnesota pioneered the strategy of double cord transplants, single cord transplants gave rise to a high incidence of graft failure and transplant related mortality. 2 Double cord transplants have now become standard when utilizing CB as the donor, as a single CB unit contains a small number of required and necessary cells for a successful transplant and double units help overcome the issues that this presents.
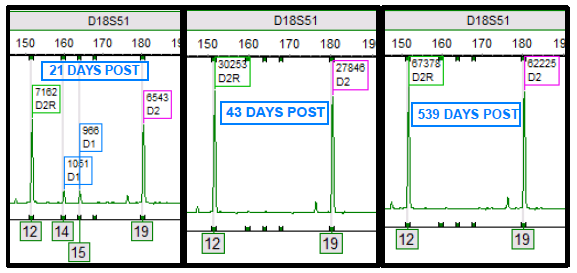
Double cord transplants are interesting and complicated for analysis purposes (and in general!). All stem cell transplants involve a dynamic process between the cells of the donor and recipient. Yet, double cords bring in another dynamic process including an additional donor.1,2 Through the chimerism monitoring process, the complexity of the engraftment process can be appreciated as one cord ultimately becomes the “winner” and the other the “loser”. In other words, one engrafts and is detectable, while the other cord fails to engraft and becomes undetectable. Figure 1 demonstrates this process, where both cords are present initially after transplant. Then, at 43 days post-transplant, a single donor cord (D2) engrafts while the other donor cord (D1) does not engraft. D1 is most likely eliminated from the host, potentially explained by multiple theories, and no longer is detectable by chimerism testing.
Figure 1. “D1” (blue) and “D2” (pink) represent donor cord one and two alleles, respectively. “D2R” (green) represent a shared allele among donor cord two and the recipient. Each image is a time lapse of the “D18S51” STR locus post-transplant. Alleles 12, 14, 15, and 19 are present at this locus. At 21 days post-transplant, both donors are present. At 43 days post-transplant and following, only donor 2 is present and alleles 14 and 15 are no longer observed.
In the case study below, the patient was diagnosed with chronic myeloid leukemia and received a double cord transplant in 2014. One would expect, as described above, that one cord would become the “winner” while the other is rejected and becomes the “loser” and becomes undetectable. Interesting enough, this patient never achieved a status of a “winner” or “loser” cord. Rather, both remained persistent within the patient’s chimerism profile and over time have become relatively stable in their percentages.
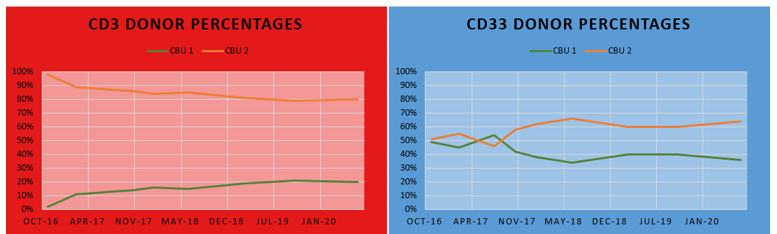
In the electropherogram below (Figure 2), alleles from both donors can be appreciated from the CD3 (top) and CD33 (bottom) lineages. Each lineage exhibits different constitutions of the donor cord percentages, where CD3 has a greater proportion of cord two than CD33; yet both lineages have a greater overall percentage of cord two than cord one. Looking at the line graph (Figure 3), the differences between the cord percentages can be further appreciated over time. It can even be noted that the cord proportions in the CD33 lineage swapped in 2017, only to swap back to favor cord two and to remain that way since. Changes of donor-recipient relative percentages occur throughout the post-transplant journey and these events are due to complex processes. Some patients become transient mixed chimerisms (who initially are mixed chimerism but later achieve total/complete chimerism), others achieve complete chimerism, and yet others may become stable mixed chimerism. It is important to note that, even in cases where complete chimerism is not achieved, disease remission can still be present.1 In this case, the patient has achieved a stable mixed chimerism status among both donor cords and, to our lab’s knowledge, is doing well clinically.
Figure 2. “D1” (blue) and “D2” (pink) represent donor cord one and two alleles, respectively. Green D1D2R, D2R, and D1D2 represent shared alleles (where “R” represents recipient alleles). Comparing the top (CD3) and bottom (CD33) electropherograms, it can be appreciated that the percentage of each cord is different for each lineage population.Figure 3. The red line graph on the left depicts the donor percentage of each cord blood unit (CBU) of CD3 lineage over time (11/2016 – 07/2020). It can be appreciated that CBU 2 is the dominant cord for CD3. The blue line graph on the left depicts the donor percentage of each CBU of CD33 lineage over time (11/2016 – 07/2020). It can be appreciated that CBU 2 is also dominant, but the differences between the cord donor percentages are much less compared to that of the CD3 lineage. Also, you can see over time that the two cords are relatively stabilizing in the percentages.
This case brings me back to a memory of my professor, who spoke briefly of this occurrence in a lecture only to quickly admit of its rarity. This is an interesting case because it represents one of those extremely uncommon instances. It is a privilege to be a part of a transplant center, like Northwestern’s, where we can witness rare and unique presentations like this. It opens up opportunities to learn and explore the complexities that transplant medicine and molecular HLA have to offer.
References
Faraci M, Bagnasco F, Leoni M, et al. Evaluation of Chimerism Dynamics after Allogeneic Hematopoietic Stem Cell Transplantation in Children with Nonmalignant Diseases. Biol Blood Marrow Transplant. 2018;24(5):1088-1093. doi:10.1016/j.bbmt.2017.12.801
Gutman JA, Riddell SR, McGoldrick S, Delaney C. Double unit cord blood transplantation: Who wins-and why do we care?. Chimerism. 2010;1(1):21-22. doi:10.4161/chim.1.1.12141
Gupta AO, Wagner JE. Umbilical Cord Blood Transplants: Current Status and Evolving Therapies. Front Pediatr. 2020;8:570282. Published 2020 Oct 2. doi:10.3389/fped.2020.570282
-Ben Dahlstrom is a recent graduate of the NorthShore University HealthSystem MLS program. He currently works as a molecular technologist for Northwestern University in their transplant lab, performing HLA typing on bone marrow and solid organ transplants. His interests include microbiology, molecular, immunology, and blood bank.
A 31 year old woman, gravida 1 para 0, 35 weeks pregnant, arrived in the emergency room via ambulance following a fall down the stairs. The ER ordered a CBC, Type and Screen and a Kleihauer-Betke (KB) test and sent blood to the lab. The KB result was positive with 1.1 % fetal cells. Hypothetically, if this was an exam question, you might be asked, “How many doses of Rhogam should be administered?” But, before you grab your calculators, let’s explore that a bit.
Hemolytic Disease of the Fetus and Newborn (HDFN) has been described since the early 1600s, before blood groups were recognized. In the early 1900s, pioneers in blood banking, Landsteiner and Weiner, discovered the ABO and Rh blood groups, and, later, the Rh system became associated with HDFN. However, the antibody related etiology and pathogenesis of HDFN was not recognized until the late 1930s. Thus, the disease was written about in memoirs of midwives and physicians as early as 1609, but the mechanism involved was not described for another 300 years. The KB test was developed in 1957 by Enno Kleihauer and Klaus Betke to quantitate fetal maternal hemorrhage (FMH). The KB test allows physicians to diagnose and monitor and to initiate therapy to prevent the effects of HDFN. Finally, considered one of the most significant successes in medicine, prophylaxis for Rh HDFN, Rh immune globulin (RhIg), became available in 1968. The KB test is used to quantitate FMH in RhD negative mothers and the results can be used to calculate dosage for RhIg to prevent immunization. The KB test became one of the earliest examples of using a laboratory test to determine the appropriate dosage of a drug.1
KB testing has traditionally been used for RhD negative women to detect FMH and to determine the appropriate dose of RhIg to prevent immunization. In an RhD negative woman, we are concerned with immunization if the baby and mother are not antigenically similar. An RhD negative mother is given a prophylactic dose of RhIg at 28 weeks gestation. After delivery, when a newborn has a positive DAT and the fetal screen is positive, a quantitative test is needed to determine the appropriate dose of RhIg. In prenatal maternal trauma, there can also be a fetal bleed. Much as in childbirth, in a trauma, the baby’s blood can enter the mother’s circulation. This indicates placental hemorrhage and can be a prediction of preterm labor. In prenatal maternal trauma, the KB test has been used as aid in diagnosis and prognosis of HDFN, preterm labor and fetal demise. It can be used to determine if there has been a fetal bleed, and if so, to determine how much RhIg should be administered.
But, did you know that the KB test can also be used to determine FMH in RhD positive mothers? This is considered an alternative usage of the test. In the labs where I did KB tests, most fetal screens in Blood Bank were held until the following morning and performed on day shift. So, any KB tests on postpartum patients were also mostly done on day shift. I worked 2nd shift, and it was not uncommon to see KB tests ordered on RhD positive women. In fact, most of the KB tests ordered on 2nd and 3rd shift were from the ER and on RhD positive mothers. With RhD positive mothers, providers are not concerned with the mother producing anti-D, so RhIg is not a concern. Therefore, the answer to the hypothetical question posed above, is that this mother did not need any RhIg because, by checking the lab results it would be noted that this woman was Rh positive with a negative antibody screen.
A study performed in 2004 at the Shock Trauma Center, University of Maryland in Baltimore, reported that pregnant trauma patients with positive KB tests often had pre term contractions All patients in their study who experienced preterm contractions had positive KB tests. None of the patients with negative KB tests had uterine contractions. The conclusion was that “Kleihauer-Betke testing accurately predicts the risk of preterm labor after maternal trauma. Clinical assessment does not.” 2 They additionally concluded that, with a negative KB test, electronic fetal monitoring could safely be reduced. The major statement of the study, which has been incorporated into practice guidelines was that KB testing is important for all pregnant trauma patients, regardless of Rh status.2,3
In 2019 the College of American Pathologists Transfusion, Apheresis and Cellular Therapy Committee sent a survey with their proficiency testing program to determine how many participating laboratories perform KB tests on Rh positive pregnant females. 52% of the labs who responded noted that they performed quantitative fetal hemoglobin testing for RhD positive women, and about 39% reported performing more than 20 tests a year. The CAP group also reviewed literature detailing 16 observational studies and concluded that the literature supporting relying on the KB as a predictor of fetal distress was lacking evidence and nonconclusive. Despite the fact that doctors are ordering these and many laboratories are still performing this test STAT on RhD positive mothers, different guidelines for practice are mixed regarding if and how the KB should be used in these RhD positive trauma patients. Furthermore, many labs responded on the survey that doctors considered these results very important but that the labs were not sure how the results helped guide management of the mother or fetus.4
One of the problems some of these guidelines cite is that the KB test may not be rapid enough to use in trauma situations. Now, I have to start by saying that KB tests are probably no tech’s favorite test. The last hospital I worked at did KB tests in Hematology. Before that I worked at a hospital where we did KB tests in Blood Bank. There seems to be no way to avoid them! I would have to agree that a KB is not at all rapid. The test is both time sensitive, always ordered STAT, and very time consuming. Hands on time is considerable. I’ve gotten 2 in one night, on 2nd shift with only 4 or 5 techs manning the whole lab, and that makes for a busy night! Add a trauma or 2 to the mix, or a few units to wash for the NICU and you know why “Kleihaur-Betke” are not our favorite words.
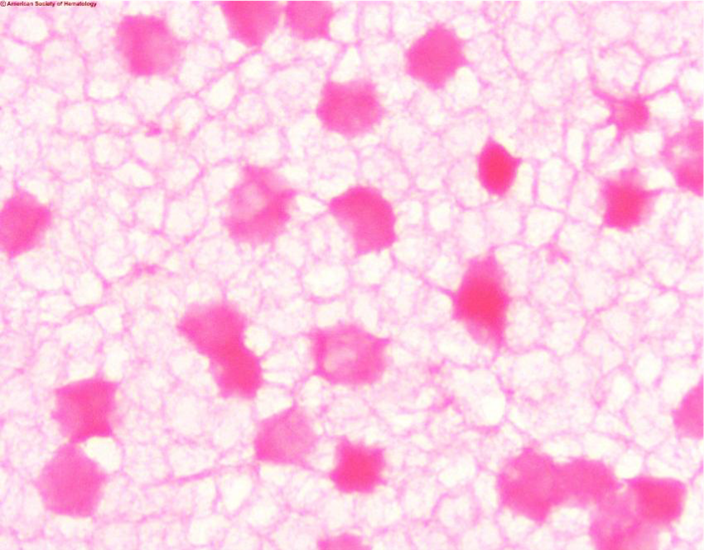
Another concern is that the KB test is marketed as a quantitative test. The problem with this is that it is not very precise due to technical difficulty. In the KB acid elution test the mother’s blood is treated with acid and then stained and counterstained. Fetal cells contain HbF which is resistant to acid and these cells will remain bright pink. The mother’s cells, which are primarily HbA, will appear as faint ‘ghost’ cells. 2000 cells are counted and the percent of fetal cells is determined. The test is complicated and needs precision in staining, counting and calculations. A slide that’s too thick, poor timing of steps, slides that are not adequately dried, or fetal cells that fail to stain can all affect results and cause false negative results. In pregnant women HbF may be increased, and in women with hemoglobinopathies such as sickle cell anemia and thalassemia Hb-F can be increased, leading to false positive results. As well, late in pregnancy it would be considered normal to have some fetal cells in the mother’s circulation. Thus, both false negative and false positive KB results are not uncommon, and a positive report on a KB test may not accurately predict fetal distress.
Image 1. Kleihauer-Betke stain showing dark ink fetal cells and ‘ghost’ like pale maternal cells
In the CAP survey article, it was noted that, of participating labs, about 96% did KB tests and 4% use flow cytometry.4 Flow cytometry is accurate, sensitive and reliable for HbF determination. Flow cytometry uses antibodies directed against fetal hemoglobin and antibodies directed against adult RBCs. A clear separation of populations can be identified and quantitated. Despite the fact that it is well known that flow cytometry is a much more precise test for FMH, many laboratories continue to do KB testing. This is likely due to the fact that only a small percentage of labs have flow cytometers. If, in trauma situations, physicians want HbF determination with a “fast” turnaround time, KB testing can be done in house with no equipment necessary. This is not fast, but would, in most circumstances, be faster than sending a test to a reference lab.
The KB test has historically been validated and used to estimate the total amount of FMH, and the results used to calculate if additional doses of RhIg are indicated. The test has high specificity for HbF but can be subjective. Precision between techs and even with the same tech repeating the test can be relatively low. Because of this, the formula used to calculate RhIg dosage has a factor built in to make up for any imprecision. An alternate usage of the test, and the one used in this case example, is to predict outcomes and guide treatment in maternal trauma victims, regardless of Rh status.
While there is some controversy on using the KB test in these cases, it is none the less still recommended by many authors and included in medical guidelines.5 Providers are using the KB test more and more for assessing placental hemorrhage in cases of trauma and premature labor. Though immunophenotyping by flow cytometry has a greater accuracy, the KB test can give reliable results at a lower cost and with a faster turnaround time.
As always, this blog led me off on several tangents while writing. When I have an idea for a blog, I start with a case study or an interesting sample I have seen in the lab. The case study itself is the easy part, then I start researching and reading articles about the disorder, test or phenomenon that I am writing about. Often, when I read one article, I ask myself another question and say, “what if…?” and that leads to another article and another and another. Days later I can still find myself reading articles and chasing after more information. I love my job, I love being a Medical Laboratory Scientist and educator, and in true form of the curious MLS, I always want to investigate and never want to stop learning. Thus, this simple case about an alternative usage of Kleihauer-Betke (KB) test kept developing as I wrote. As a side note, it was interesting to see that the studies have had different conclusions and the guidelines for this use of the KB test have swayed over the years. It will be interesting to see what the future will bring. I have seen some articles about adding the HbF determination to hematology analyzers—wouldn’t that be nice!
References
Reali G. Forty years of anti-D immunoprophylaxis. Blood Transfus. 2007;5(1):3-6. doi:10.2450/2007.0b18-06
Muench MV, Baschat AA, Reddy UM, Mighty HE, Weiner CP, Scalea TM, et al. Kleinhauer-betke testing is important in all cases of maternal trauma. J Trauma 2004;57(5):1094-8.
Michael V. Muench, Joseph C. Canterino, Trauma in Pregnancy, Obstetrics and Gynecology Clinics of North America, Volume 34, Issue 3, 2007, Pages 555-583.
Matthew S. Karafin, Chad Glisch, et al, for the College of American Pathologists, Transfusion, Apheresis, and Cellular Therapy Committee; Use of Fetal Hemoglobin Quantitation for Rh-Positive Pregnant Females: A National Survey and Review of the Literature. Arch Pathol Lab Med 1 December 2019; 143 (12): 1539–1544.
-Becky Socha, MS, MLS(ASCP)CM BB CM graduated from Merrimack College in N. Andover, Massachusetts with a BS in Medical Technology and completed her MS in Clinical Laboratory Sciences at the University of Massachusetts, Lowell. She has worked as a Medical Technologist for over 30 years. She’s worked in all areas of the clinical laboratory, but has a special interest in Hematology and Blood Banking. When she’s not busy being a mad scientist, she can be found outside riding her bicycle.
Since 2008 I have served as the Associate Medical Director and the Medical Director of Transfusion Medicine in a large academic medical center. In addition to overseeing the operations of our transfusion service, I also spend several days per week in our apheresis unit. We currently see between 10-20 patients daily for a wide range of therapeutic apheresis procedures performed by our 5 apheresis nurses including stem cell collection and lymphapheresis procedures for stem cell transplant and CarTcell therapy, respectively. These procedures can last from 90 minutes to 6 hours and includes both outpatients as well as acutely ill patients in our critical care units. Typically we perform procedures from 8 AM to 6 PM but there are frequent requests for procedures that last beyond these hours and occasionally in the middle of the night for life threatening conditions. Despite the long hours and unpredictable days, this provides an opportunity to bond with patients over the hours and days they spend in our apheresis unit.
I remember the first time I met Reed. He was sitting on the side of his hospital bed and was bald and pale in stark contrast to his dark blue pajamas. Although he was thin, I could tell that he was a much bigger man before chemotherapy and the transplant ravaged his body. I introduced myself and he was pleasant and engaging despite how ill he was. He had recently undergone 2 autologous stem cell transplants and now with recurrence of the multiple myeloma he had received his brother’s stem cells and was suffering severe acute graft vs. host disease (GVHD). His entire gastrointestinal (GI) tract was under assault as he was diagnosed with Grade IV GI GVHD and was losing liters of bloody stool daily. Despite the abdominal pain and cramps, I never saw him without a smile on his face. He had been treated with high doses of immunosuppression but his GVHD was unresponsive and now we were called in to perform photopheresis, which has great results for skin and pulmonary GVHD but has not been as effective for GI GVHD. In fact, all our previous patients with Grade IV GI GVHD lost their battle.
The bone marrow transplant physician advised Reed that his prognosis was poor and that he should get his affairs in order. His response to the BMT physician was, I am not leaving my wife to raise our three children by herself and I am going to walk out of this hospital. We performed photopheresis twice a week every week and gradually his symptoms improved. His hair started growing back, his color returned, and he kept his word and walked out of the hospital. He continued photopheresis twice a week every two weeks for 2 years. During that time, he met my son who was only 8 at the time and I met his wife and children. He always asked how my son was every time he came for his treatment and what activities he was involved in. When he finished his 2 years of photopheresis, he brought every pathologist and nurse a long stem red rose and thanked us for saving his life.
Several years later, Reed started to experience renal failure as another complication of GVHD and again he was referred to our clinic for plasmapheresis. We picked up where we left off during his weekly treatments. Again, his positive attitude and compliance with treatment were successful in saving his kidneys. This past summer I went to an outdoor concert. At the end of the night, when everyone was leaving, I saw Reed and his wife! I was so happy to see him looking healthy and strong. I introduced him to everyone who was with me, telling them that Reed was our miracle patient, the only patient that survived Grade IV GI GVHD. This fall, a card was delivered to my office. It was a birthday card from Reed to celebrate my 50th birthday! That is so typical of Reed, still thinking about others and wanting to do what he can to show how important others are to him!!
-Kimberly Sanford, MD is the Medical Director of Transfusion Medicine at Virginia Commonwealth University Health.
When I first thought of writing a blog on blood supplies amid the COVID-19 pandemic, it was early March. Fast forward a couple months and a lot of things have changed. So, where were we, and where are we now?
January 6, 2020
At this time, most people in the US were not even aware of the novel coronavirus. (unless you were taking my Introduction to Human Disease course and were searching online for media articles about infectious disease!)
I first became aware of this ‘mystery’ virus in early January, when I was teaching an online Winter session course called Introduction to Human Disease. I developed this course a number of years ago as a STEM course for non-science majors. The intent of the course is to familiarize students with diseases and disease terminology that they will use in their everyday lives. The course gives students a chance to learn basic medical concepts that will enable them to become their own (or their family’s) medical advocate. In addition, the course covers many diseases that are ‘in the news’ and allows students to gain some knowledge and insight into the myths and facts surrounding these diseases. Topics covered include general mechanisms of disease, including inflammation, infectious disease, immunity, heredity, and cancer. Emphasis is placed on emerging and pandemic …. so, when this disease emerged, we were right there to take note!
I asked the students to find an article in the media on an infectious disease, and to summarize and answer questions about the article and the mechanism of the disease. Three students chose different articles about this yet unnamed mystery illness affecting people in Wuhan, China. We had active discussion board conversations about this emerging severe respiratory disease and pneumonia, that at the time had infected around 40 people, with no reported deaths, and no human to human transmission. In my comments, I compared this novel virus to seasonal influenza, H1N1, SARS and MERS and tried to reassure students that this would hopefully follow the same path as H1N1 or SARS and MERS.
Feb 21, 2020
The first confirmed case in the United States was on Jan 21 in Washington state. (CDC)1 On Jan 31, the Health and Human Services Secretary declared Coronavirus a Public Health Emergency in the US. (HHS.gov)2 We began hearing news of restrictions on flights from China, passengers affected on Princess Cruise ships and outbreaks at a long term care facility in Washington State.
As a Medical laboratory Scientist, I became concerned with this virus early on, and started watching statistics. I was concerned not only for the health of my family, friends and coworkers, but also for the health or our laboratories and our blood supply.
The first journal articles I read about COVID-19 and blood safety were published in Transfusion Medicine Reviews on Feb 21, 2020. In the very early days of this novel coronavirus, researchers in China reviewed publications about SARS and MERS to help give us a better understanding of SARS-CoV-2, the virus that causes our current pandemic of COVID-19. When discussing blood safety, one of the first things to consider is if the virus is transmittable via blood transfusions. If the virus is transmittable, we also must consider if there is an asymptomatic time when there is virus in the blood. One review stated that SARS, MERS and SARS-CoV-2 can all be found in the serum or plasma, but, at the time of this review, it was still uncertain if SARS-CoV-2 could be transmitted from those with pre-symptomatic or asymptomatic infections.3
March 18, 2020
On March 18, Blood Transfusion published an article written by a group at several Blood Centers in a few provinces in China. This article discussed efforts to minimize the impact of blood shortages due to COVID-19. It was noted that the rising pandemic had had a profound impact on the number of blood donations, and on blood safety. Because it was now recognized that there is a long incubation period and a significant number of asymptomatic cases, this posed a huge challenge in recruiting blood donors. In China, strictly restricted mobility led to a decrease in donations across the country. Donors were recruited through various methods, including the use of social media. Social distancing during blood donations and thorough cleaning and disinfecting of donor areas were enforced. Screening procedures were enhanced to include temporary isolation of blood products for 14 days after collection and delaying release for clinical use. At the same time, donors were followed up until the expiration of the products. If a donor was found to test positive for COVID-19 after donation, the blood products were recalled. These new protocols in place were helping to insure adequate donations and the safety of blood products. ne interesting note is that this article referred to the epidemic as “effectively controlled” and that “normal medical services had been resumed”.4
Meanwhile, in the US, American Red Cross was pleading for blood donors. On March 17 it was reported that 2,700 mobile blood drives had been cancelled at a loss of 86,000 units of blood potentially collected. On March 21, 4 days later, that number had risen to more than 5,000 blood drives canceled at a loss of 170,000 units. As more schools, workplaces, churches and college campuses closed down in response to the pandemic, those institutions had to cancel their blood drives. Social distancing guidelines and shelter in place orders resulted in fewer people donating blood. In addition, an FDA mandate from February, that people who had traveled to areas with COVID-19 outbreaks should wait at least 28 days before donating blood, most likely contributed to the shortage. Dr. Justin Kreuter, from the Mayo Clinic Blood Donor Center, stated that the blood shortage was not due to more COVID-19 patients needing blood products. Rather, “it’s a lack of donations coming in.”5
April 1, 2020
procedures that the Chinese had instated. Mobile blood drives were shut down, but collection centers remained open. TV commercials, radio ads, You Tube videos and social media called for blood donors, assuring them that this was essential and that donating blood was safe. Donations were arranged through appointments only, and potential donors contacted and verbally screened for symptoms and risk factors before appearing to donate. On arrival at the centers, temperatures were taken and travel and symptoms questions were asked before a donor was allowed to enter the center. The use of masks and social distancing, along with extra cleaning and donor chair decontamination between donors were all implemented.
In an effort to open up the pool of potential donors, the FDA reviewed current studies and epidemiological data and concluded that certain donor eligibility criteria could be modified without compromising the safety of the blood supply. On April 2, 2020 the FDA approved several important changes in donor qualifications. These revisions included the following:
For male donors deferred for having sex with another male: the recommended deferral period changed from 12 months to 3 months.
For female donors deferred for having sex with a man who had sex with another man: the recommended deferral period changed from 12 months to 3 months
The deferral period for recent tattoos and piercing was changed from 12 months to 3 months
For people who have traveled to malaria-endemic areas, the recommended deferral period was changed from 12 months to 3 months. In addition, the guidance notes that deferral can be waived for these donors, provided the blood components are pathogen-reduced using an FDA-approved pathogen reduction device.
For donors who spent time in European countries or on military bases in Europe who were previously deferred due to potential risk of transmission of Creutzfeldt-Jakob Disease or Variant Creutzfeldt-Jakob Disease, the FDA has eliminated the deferrals and these individuals may now qualify to donate.6
Despite loosening requirements, advertising, and calls from the blood centers for additional donors, the shortages remained. To address the decline in blood product availability, it became essential to review the principles of patient blood management (PBM). PBM is defined as “the timely application of evidence-based medical and surgical concepts designed to maintain hemoglobin concentration, optimize hemostasis and minimize blood loss in an effort to improve patient outcome.”7 Firstly, elective procedures were put on hold, thus freeing up units for the most needy patients. Despite this, many blood banks still had their standing orders decreased. In many cases, Blood bank Medical Directors approved changes in transfusion triggers. At the hospital where I work, the transfusion trigger was changed from a hemoglobin of 8g/dL to 7 g/dL. New changes of SOP were approved to issue to all patients, except females of child bearing age, Rh positive units instead of more scarce Rh negative units. We also have a large NICU unit and baby units were not available from ARC, so we were using the newest units available, when necessary for these patients.
By April 8, 15,000 blood drives had been cancelled across the US, at a potential loss of almost 500,000 donated units. One technologist reported in an online Blood bank professionals group, that “Our supplier downgraded us in terms of standard inventory (about 40%), but our transfusion numbers have dropped at least as much.”8 With the decrease in usage and the careful patient blood management, blood needs were met.
May 12, 2020
AABB began sending out a weekly COVID impact survey for hospital transfusion services survey in late March. Many questions on the survey, and the resulting charts and graphs, are related to COVID convalescent plasma practices and procedures (details in my next blog!), but one important graph produced by this survey shows the increase in inventory wastage due to changes related to COVID-19. These changes due to COVID-19 can be a decrease in patients and elective surgeries or changes in transfusion protocols. In early April, in the first few weeks of the survey, 25%-28% of hospitals responding reported an increase in inventory wastage. This corresponds to when donors started coming back to donate, and usage dropped. This percent of hospitals reporting wastage increased each week until the week of May 4-7 when 54% of hospitals reported inventory wastage. This may be due to several factors. The units collected at the end of March and early April, have reached their 42 day expirations. Donors came out initially in response to the call for blood, but now, these units have expired, and it has not yet been 56 days when these donors can donate again. Usage also decreased during this time. COVID patients have not generally had heavy use of red cells, in particular, and doctors have been very conservative in usage with all patients. For the week of May 11-14, as more hospitals are planning to resume elective surgeries, and for the first time in the 8 weeks, fewer hospitals (52.0%) reported an increase in wastage due to changes related to the pandemic. Of the 100 respondents, 59% reported they are resuming “some” elective surgeries before mid-May and 28.0% are doing so after mid-May.9
What does this mean for the future of our blood supply during this pandemic? On May 12, a group of Blood bank professionals, when asked in an informal online survey, had had varying answers. These were likely dependent on location, both geographic and city vs. rural, and size of the hospital. One comment was that “We have gone from huge shortages to throwing away massive units not being used. Hospital is empty.” Another tech said “We were way overstocked a week ago, now we’re dipping way below average.” Technologists in Florida, Oregon and Pennsylvania reported low inventory. Techs in Ohio and Maryland reported their inventory to be very healthy. But these reports could easily vary between areas of the individual state, and even different hospitals in the same city. Another technologist commented “We had a mass of donors when this all started and now all those units are expiring!” The shortage of donors will likely continue, but may relax a bit with some states beginning to lift restrictions. We likely won’t see a huge drove of donors, all at once, which is actually good because it will spread out expiration dates. But, though things may be opening up, it is unlikely that we will see blood drives at schools, workplaces and churches for some time, and this is a huge source of our countries blood supply.
We have seen a big swing in both inventories and usage. After elective and with surgeries have been put on hold for months, we may see an increase over the typical number of elective surgeries, which will mean we will see an increase in blood usage, and with a lack of donors, inventories may drop again.
As far as blood safety, we know now that SARS-CoV-2 did not follow the path of SARS and MERS. We know that it can definitely be transmitted from person to person, and can be transmitted by people who are asymptomatic. But, we also know that, in general, respiratory viruses are not known to be transmitted by blood transfusion. So, from what we know at this time, it is likely not necessary to routinely screen blood products for SARS-CoV-2, and not necessary to isolate blood products after collection and delay release of the products. It is recommended that blood centers encourage self-deferral for donors who have traveled to a COVID-19 affected area or been in contact with an infected person in the past 14 days and to screen donors carefully for fever and respiratory symptoms. With these practices in place, we can ensure an adequate and safe blood supply. We will continue to see swings in volumes, but with careful patient blood management, we will ride these waves and come out on top. Thanks to all our wonderful Blood Bank Technologists who are helping manage our country’s blood supplies!
-Becky Socha, MS, MLS(ASCP)CM BB CM graduated from Merrimack College in N. Andover, Massachusetts with a BS in Medical Technology and completed her MS in Clinical Laboratory Sciences at the University of Massachusetts, Lowell. She has worked as a Medical Technologist for over 30 years. She’s worked in all areas of the clinical laboratory, but has a special interest in Hematology and Blood Banking. When she’s not busy being a mad scientist, she can be found outside riding her bicycle.