A middle-aged male with a history of type 2 diabetes, hypertension, obesity, and alcohol-related cirrhosis presented to the emergency department with altered mental status. He was obtunded, acutely encephalopathic and hypoglycemic. He soon developed emesis and coded during clinical assessment, undergoing emergent intubation. He was found to be profoundly acidotic with labs consistent with disseminated intravascular coagulation and multiorgan failure. The patient was transfused but continued to code multiple times before and during ICU transfer/admission. Despite multiple resuscitation attempts, he expired soon afterwords.
Laboratory workup
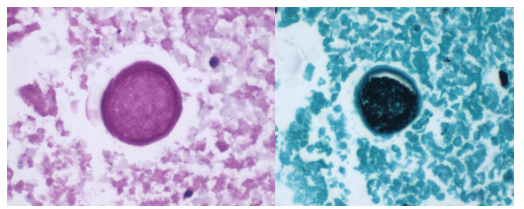
Blood cultures drawn in the ED prior to admission became positive with curved gram-negative rods (Image 1A) within 16 hours. An oxidase-positive, indole-positive, beta-hemolytic organism was recovered after 24 hours of incubation. The organism was lactose-fermenting (confirmed by ONPG) and exhibited colorless growth on Thiosulfate Citrate Bile Salts agar suggestive of a lack of sucrose utilization (Image 1B, set up for demonstrative purposes). The organism was definitively identified as Vibrio vulnificus by MALDI-TOF MS. No additional workup was undertaken as the patient had expired prior to the organism being recovered from blood culture.
Image 1. A: Gram stain from a positive blood bottle revealing curve gram-negative rods (100X magnification). Inset demonstrates the morphologically distinct curved appearance. B: Blood and TCBS agars revealing growth of V. vulnificus. Lack of yellow colorization on TCBS media indicates lack of sucrose utilization. Biochemical testing revealed oxidase, catalase, and indole positivity also consistent with the MALDI-TOF identification of V. vulnificius.
Discussion
Vibrio sp. are marine bacteria that naturally colonize brackish and saltwater aquatic environments. Of the more than 70 currently recognized species, at least 12 are recognized as human pathogens.1 Human infections are broadly classified as being either cholera (caused by V. cholerae) or vibriosis (caused by other non-V. cholerae Vibrio spp.). Unlike cholera; a severe diarrheal illness usually acquired through ingestion of contaminated food or water, vibriosis represents a group of infections with varied clinical manifestations dependent upon the etiologic agent, route of infection, and host susceptibility.2 Non-cholera vibrios are often found in seawater with moderate to high salinity and as clinically important contaminants of raw or undercooked seafood.
V. vulnificus thrives in warmer water and infections follow a seasonality, peaking in the warmer summer months.3 In contrast to V. cholerae and V. parahaemolyticus, V. vulnificus infections generally are associated with patients with underlying conditions, most commonly diabetes, liver disease and iron storage disorders. It is estimated that patients with chronic liver diseases (particularly cirrhosis due to either alcoholism or chronic hepatitis B or C) are 80-fold more likely to develop V. vulnificus-associated primary septicemia than healthy counterparts.2 Infections are most common in men aged 45-60 years who make up 85-90% of patients,4 consistent with this case. Contaminated food consumption (particularly filter feeding shellfish) can result in gastroenteritis or primary septicemia and disseminated disease.
Non-cholera vibrios are estimated to cause up to 80,000 infections worldwide, with V. parahaemolyicus and V. alginolyticus responsible for most cases. Among this group of organisms, V. vulnificus stands out as being particularly virulent; between 150-200 V. vulnificus infections are reported to the US CDC annually, with 20% being fatal.5 This organism oftencauses more than 95% of seafood-related deaths in the United States and the highest case fatality of any foodborne pathogen [2]. V. vulnificus also causes serious skin/soft tissue infections. This can occur either through exposure of a preexisting wound to contaminated seawater, or through injury while handling contaminated seafood. Cutaneous infections can present as cellulitis or bullae, which can progress to necrotizing disease and secondary sepsis is left untreated.6
No epidemiological link was able to be established between this patient’s case and either seafood or exposure to seawater, although cases of V. vulnificus infections among patients without classical exposure risk has been documented.7 This case of V. vulnificus primary septicemia highlights the acuity of this presentation as well as the importance of including V. vulnificus in differential diagnosis of hosts with associated risk factors and/or epidemiological links. Blood culture remains the gold standard for diagnosis. Importantly, doxycycline in combination with a third-generation cephalosporin constitutes the standard regimen for antibiotic therapy – however, doxycycline is not usually considered a first-line antibiotic for management of patient with gram-negative bloodstream infections, so rapid and accurate identification of V. vulnificus in this setting is essential.
1. Kokashvili, T., et al., Occurrence and Diversity of Clinically Important Vibrio Species in the Aquatic Environment of Georgia. Front Public Health, 2015. 3: p. 232.
2. Baker-Austin, C., et al., Vibrio spp. infections. Nature Reviews Disease Primers, 2018. 4(1): p. 1-19.
3. Hughes, M.J., et al., Notes from the Field: Severe Vibrio vulnificus Infections During Heat Waves – Three Eastern U.S. States, July-August 2023. MMWR Morb Mortal Wkly Rep, 2024. 73(4): p. 84-85.
4. Jones, M.K. and J.D. Oliver, Vibrio vulnificus: disease and pathogenesis. Infect Immun, 2009. 77(5): p. 1723-33.
5. Bharathan, A., et al., Implication of environmental factors on the pathogenicity of Vibrio vulnificus: Insights into gene activation and disease outbreak. Microb Pathog, 2025. 204: p. 107591.
6. Coerdt, K.M. and A. Khachemoune, Vibrio vulnificus: Review of Mild to Life-threatening Skin Infections. Cutis, 2021. 107(2): p. E12-e17.
7. Candelli, M., et al., Vibrio vulnificus—A Review with a Special Focus on Sepsis. Microorganisms, 2025. 13(1): p. 128.
-Rene Bulnes, MD is an Infectious Diseases Clinician and current Medical Microbiology Fellow at the University of Texas Southwestern Medical enter in Dallas, TX.
-Andrew Clark, PhD, D(ABMM) is an Assistant Professor at the Johns Hopkins University School of Medicine in the Department of Pathology, and Director of the Bacteriology Laboratory at the Johns Hopkins Hospital. He completed a CPEP-accredited postdoctoral fellowship in Medical and Public Health Microbiology at National Institutes of Health, and is interested in the molecular mechanisms of antimicrobial resistance, susceptibility testing, and the evaluation of novel technology for the clinical microbiology laboratory.
A-58-year-old female with past medical history of breast cancer in remission, renal transplant 8 years ago on tacrolimus, now presenting with inability to tolerate oral intake, dyspnea on exertion and gastrointestinal symptoms such as profuse foul-smelling, non-bloody diarrhea, and vomiting. She denied exposure to sick contacts, recent travel, or changes in diet. She had other co-morbidities including hypertension, atrial arrhythmia, hyperlipidemia, and past C. difficile infection. On the physical exam, she was afebrile, normotensive, with a normal heart rate and rhythm, and her abdomen is soft and non-distended, with tenderness to palpation. Cardiology was consulted due to new onset paroxysmal atrial fibrillation on telemetry, and the renal transplant team was contacted for admission in concern for her rising creatinine. Upon admission to the floor, the patient had 2 more loose stools which were collected for stool culture and multiplex, syndromic gastrointestinal PCR panel, which tested positive for norovirus. She was kept on contact precautions.
Discussion
Norovirus is a single stranded positive-sense RNA virus belonging to the Calciciviridae family. Noroviruses are divided into 10 genogroups based on the amino acid sequence of VP1, the norovirus capsid protein. Genogroups GI and GII account for 90% of reported infections including outbreaks, with the GII.4 genotype being the cause of most severe disease, and it is more frequently implicated in outbreaks than other genotypes (1).
The median incubation period for norovirus infections is 1.2 days, with most symptomatic patients presenting with diarrhea and vomiting. Asymptomatic shedding of norovirus is common, mainly in the pediatric population, where 11.6 – 49.2% of stool samples were found to contain norovirus in random recruitment studies globally. Most patients with norovirus infection have spontaneous resolution of symptoms by the third day of illness. Elderly, young, and immunocompromised patients face greater risk of severe and prolonged symptoms from norovirus infection. Chronic infection with norovirus may occur in immunocompromised hosts, with associated symptoms lasting up to weeks or months in these patients (2).
Transmission of norovirus occurs through oral-oral and fecal-oral routes, and transmission can occur directly via exposure to human emesis or feces, or through contamination of food, water, or fomites with such samples. Median viral titers have been recorded at 3.9 x 104 copies/mL in emesis samples from patients with norovirus GII, with aerosolization of viral particles possible due to projectile vomiting and toilet flushing (de Graaf). Once inoculated, norovirus particles primarily infect macrophages and dendritic cells in the gastrointestinal tract, with current reports suggesting that particles enter these cells via attachment to blood group antigens, or HGBAs, and Toll-like receptors (3). VP1, the capsid protein of norovirus, has been found to induce expression of aquaporin-1 in human intestinal cell culture. This leads to small molecule permeability at the intestinal barrier, which likely leads to the watery diarrhea seen in norovirus infection (4).
Norovirus is implicated as the cause of 19% of all acute gastroenteritis cases globally, with virtually every country reporting norovirus cases (5). These infections frequently occur in the form of outbreaks, with the highest rates of infection recorded in the winter months (Figure 1). Crowded and closed environments, including daycare centers, cruise ships, and restaurants, are known facilitators of outbreaks. More than half of all norovirus outbreaks are reported in healthcare settings, such as hospitals and long-term care facilities (6).
Immunological assays (e.g., antigen detection) and transmission electron microscopy can be used for detection of gastrointestinal viruses, but these methods have limited sensitivity and specificity and not recommended for clinical diagnosis (7). Laboratory detection by molecular methods is the preferred method for norovirus diagnosis. Testing of stool specimens may be performed on single plex or FDA-approved, commercially available, multiplex syndromic PCR panels. While PCR is the most sensitive approach, false-positives have been reported (8). Five regions of the genome (A,B,C,D, and E) of the norovirus genome have been used for genotyping while viral capsid gene (encoded by regions C, D, and E) is typically used given the viral capsid being involved in host-receptor interactions and immune response (9). In certain instances, sequencing and detection of the ORF1 and ORF2 genes may help identify strains after antigenic drift events (10).
Oral or intravenous rehydration is the mainstay of norovirus treatment in all patients. In immunocompetent patients, norovirus is expected to resolve spontaneously (11). Chronic symptomatic infection with norovirus in immunocompromised patients poses a significant clinical challenge, especially when reduction in immunosuppressants is not feasible. In such patients, case reports have suggested that nitazoxanide may achieve resolution of symptoms, and one retrospective study proposed that addition of metronidazole led to resolution of norovirus symptoms in nitazoxanide-refractory cases (12-14). However, no randomized controlled trials have demonstrated the efficacy of nitazoxanide or metronidazole in chronic norovirus infection. Norovirus vaccines are currently in development, but challenges include high genetic diversity of circulating strains, lack of understanding regarding herd immunity and correlates of immune response, and the lack of standardized testing approaches such as cell cultures or animal models for efficacy studies.
References
1. Carlson KB, Dilley A, O’Grady T, Johnson JA, Lopman B, Viscidi E. A narrative review of norovirus epidemiology, biology, and challenges to vaccine development. NPJ Vaccines. 2024;9(1):94. Published 2024 May 29. doi:10.1038/s41541-024-00884-2
3. Chen J, Cheng Z, Chen J, Qian L, Wang H, Liu Y. Advances in human norovirus research: Vaccines, genotype distribution and antiviral strategies. Virus Res. 2024;350:199486. doi:10.1016/j.virusres.2024.199486
4. Zhang M, Zhang B, Chen R, et al. Human Norovirus Induces Aquaporin 1 Production by Activating NF-κB Signaling Pathway. Viruses. 2022;14(4):842. Published 2022 Apr 18. doi:10.3390/v14040842
5. Zhang P, Hao C, Di X, et al. Global prevalence of norovirus gastroenteritis after emergence of the GII.4 Sydney 2012 variant: a systematic review and meta-analysis. Front Public Health. 2024;12:1373322. Published 2024 Jun 27. doi:10.3389/fpubh.2024.1373322
6. Tsai H, Yune P, Rao M. Norovirus disease among older adults. Ther Adv Infect Dis. 2022;9:20499361221136760. Published 2022 Nov 14. doi:10.1177/20499361221136760
7. Rabenau HF, Stürmer M, Buxbaum S, Walczok A, Preiser W, Doerr HW. Laboratory diagnosis of norovirus: which method is the best? Intervirology. 2003;46(4):232-8. doi: 10.1159/000072433.
8. Caza M, Kuchinski K, Locher K, Gubbay J, Harms M, Goldfarb DM, Floyd R, Kenmuir E, Kalhor M, Charles M, Prystajecky N, Wilmer A. Investigation of suspected false positive norovirus results on a syndromic gastrointestinal multiplex molecular panel. J Clin Virol. 2024 Dec;175:105732. doi: 10.1016/j.jcv.2024.105732. Epub 2024 Sep 30.
9. Mattison K, Grudeski E, Auk B, Brassard J, Charest H, Dust K, Gubbay J, Hatchette TF, Houde A, Jean J, Jones T, Lee BE, Mamiya H, McDonald R, Mykytczuk O, Pang X, Petrich A, Plante D, Ritchie G, Wong J, Booth TF. Analytical performance of norovirus real-time RT-PCR detection protocols in Canadian laboratories. J Clin Virol. 2011 Feb;50(2):109-13. doi: 10.1016/j.jcv.2010.10.008. Epub 2010 Nov 10.
10. Mattison K, Grudeski E, Auk B, Charest H, Drews SJ, Fritzinger A, Gregoricus N, Hayward S, Houde A, Lee BE, Pang XL, Wong J, Booth TF, Vinjé J. Multicenter comparison of two norovirus ORF2-based genotyping protocols. J Clin Microbiol. 2009 Dec;47(12):3927-32. doi: 10.1128/JCM.00497-09. Epub 2009 Oct 21.
12. Haubrich K, Gantt S, Blydt-Hansen T. Successful treatment of chronic norovirus gastroenteritis with nitazoxanide in a pediatric kidney transplant recipient. Pediatr Transplant. 2018;22(4):e13186. doi:10.1111/petr.13186
14. Soneji M, Newman AM, Toia J, Muller WJ. Metronidazole for treatment of norovirus in pediatric transplant recipients. Pediatr Transplant. 2022;26(8):e14390. doi:10.1111/petr.14390
-Brendan Sweeney is a third-year medical student at the George Washington University School of Medicine and Health Sciences. His research interests include infectious diseases, hematopathology, and point of care diagnostics.
-Rebecca Yee, PhD, D(ABMM), M(ASCP)CM is the Chief of Microbiology, Director of Clinical Microbiology and Molecular Microbiology Laboratory at the George Washington University Hospital. Her interests include bacteriology, antimicrobial resistance, and development of infectious disease diagnostics.
An 11-year-old girl was brought by her mother to the Emergency Room because of altered mental status, described as abnormal movements and staring with a period unresponsiveness lasting 15 minutes.
One week previously, she had returned from a 6 week trip to Ghana with her family to visit other family members there. Malaria prophylaxis had been prescribed but was not taken during the trip. Since coming back, she had constant frontal and bilateral headaches with retro-orbital pain, accompanied by nausea, poor appetite, and several episodes of vomiting. She did not have cough, congestion, earaches, or fever. Past medical history was unremarkable. Nobody else in the family was sick. She had some mostquito bites while in Ghana, but no fever or illness.Confusion, fever, and neck stiffness were noted on physical exam.
A spinal tap was done, with the following results: wbc 46 per mm3 (lymphocytes 98%, monocytes 2%), rbc 25 per mm3, glu 41 mg/dL, pro 72 mg/dL, no organisms on Gram stain and Kinyoun stains, meningitis/encephalitis PCR panel negative, and PCR for Mycobacterium tuberculosis negative. The bacterial culture had no growth after 5 days of incubation.
A CT scan of the head done without contrast was normal. An MRI of the brain done with contrast showed focal contrast enhancement in the left corona radiata and several other small foci of contrast enhancement, including within the right occipital lobe and cerebellum, alsong with possible leptomeningeal contrast enhancement along several sulci.
After 4 weeks, growth was observed in the Mycobacterial Growth Indicator Tube (MGIT) culture. A Kinyoun stain of the growth is shown in Figure 1, and colony morphology is shown in Figure 2. The organism was subcultured on the Middlebrook 7H10 (the growth shown in Figure 2) and identified by MALDI-ToF (Matrix assisted laser desorption ionization Time of Flight) as Mycobacterium tuberculosis complex (MTBc). The antimicrobial susceptibility test was performed at the department of health, which reported out susceptible to all first-line agents, except resistance to INH.
Fig 1 (A): Acid-fast bacilli from Kinyon stain of positive MGIT culture.Fig 1(B and C): Close up images of Fig1-A.Fig 2: Dry Crusty scaly morphology of Mycobacteria subcultured from positive MGIT.
Discussion
Tuberculous (TB) meningitis is a severe form of extrapulmonary tuberculosis caused by Mycobacterium tuberculosis (Mtb). It typically presents with a subacute onset of constitutional symptoms, including malaise, fever, headache, and altered mental status, which can progress to stupor, coma, and death if untreated. Clinical features often include headache, vomiting, meningeal signs, focal neurological deficits, cranial nerve palsies (our case has cranial nerve 6 palsy), and raised intracranial pressure.
The diagnosis of TB meningitis is generally based on clinical suspicion, CSF analysis, and neuroimaging. CSF analysis is typically non-specific and shows lymphocytic pleocytosis, elevated protein, and low glucose levels. Confirmatory tests include CSF smear, culture, and nucleic acid amplification tests for Mtb. While mycobacterial culture is still a gold-standard method for the definitive diagnosis, it usually takes long for growth detection and the downstream diagnostic methods, such as MALDI-ToF (Matrix Assisted Laser Desorption Ionization Time of Flight). Since laboratories are reliant on (MALDI-ToF) after the discontinuation of Hologic GenProbe products, subculturing the organism from the liquid growth for MALDI-ToF results in additional delay in identification.
The cording characteristics of MTB from culture growth was a classic tell-tale sign for preliminary laboratory identification. While the presence of “cord factor” denotes the virulence of mycobacterial species (particularly MTBc) and was thought to be unique to MTBC, it was later demonstrated to be present in non-tuberculous mycobacterial (NTM) species. Therefore, care should be taken, and the time of growth should be considered when interpreting the Kinyon stain of positive cultures.
On the other hand, Xpert MTB/RIF is FDA-approved only on sputum samples, although studies show off-label utilization on CSF. The sensitivity of this test in CSF is mediocre due to the paucibacillary nature of the infection. Neuroimaging, such as MRI or CT, can reveal meningeal enhancement and hydrocephalus, which are suggestive of TB meningitis; however, clinicians still rely heavily on microbiologic results for definitive diagnosis.
As TB meningitis is a fatal disease and the confirmed diagnosis may take a long time, treatment should be initiated promptly based on clinical suspicions. Treatment includes anti-tuberculous medications with steroids for 2 months. Then NIH and RIF for an additional 7-10 months. It is noteworthy to mention that susceptibility is important as some MTB strains are drug-resistant, as is the case for our patient, whose isolate is resistant to INH.
Theorn et al. Scientific Reports. 2014. DOI: 10.1038/srep05658. Accessed. June 19, 2024
Wilhelm Hedin et al., JID, 2023
Lablogatory – a cording too cording by Richard Davis
-Dr. Mahmoud Ali, MD, is a pediatric infectious disease fellow at Montefiore Medical Center and Albert Einstein College of Medicine, Bronx, NY.
-Phyu Thwe, Ph.D, D(ABMM), MLS(ASCP)CM is Associate Director of Infectious Disease Testing Laboratory at Montefiore Medical Center, Bronx, NY. She completed her medical and public health microbiology fellowship in University of Texas Medical Branch (UTMB), Galveston, TX. Her interests includes appropriate test utilization, diagnostic stewardship, development of molecular infectious disease testing, and extrapulmonary tuberculosis.
Case 1: Premature newborn with abnormal reticulocyte scattergram, indicated by the * after the parameters. The sample was rerun with a 1:5 dilution to minimize interference. CBC and Retic results shown below.
Table 1. newborn CBC results before and after 1:5 dilution
In this case the dilution was able to minimize interference, so there is no longer an abnormal scattergram flag. After diluting the sample, the HCT was multiplied by the dilution factor. The MCV, Reticulocyte % and Immature Reticulocyte Fraction (IRF) are not multiplied because these are % and fractions or ratios so do not change.
Case 2: 48-year-old woman with sickle cell anemia with abnormal reticulocyte scattergram. The sample was rerun with a 1:3 dilution to minimize interference. CBC and reticulocyte results are shown below.
Table 2. Sickle cell patient results before and after 1:3 dilution
In this case, there is still an * after the reticulocyte % and Immature Reticulocyte Fraction (IRF) indicating that the dilution did not help minimize the interference in the scattergram. In Case 1 a 1:5 dilution was used. In this sickle cell patient, the red blood cell (RBC) count is too low to do a higher dilution. (The raw RBC with the 1:3 dilution was .69. On our analyzer it is important not to dilute a sample so that the RBC would be <0.5 as this suppresses the analyzer flags) This means that reticulocyte results could not be reported from the analyzer, and it was necessary to verify the reticulocyte count with a manual reticulocyte count.
I’ve often said that I really love working in Hematology, especially when these problem specimens come along. About 85% of our CBC’s autovalidate, so it’s easy to say that those aren’t too interesting. It’s the ones that need our attention that that are interesting and give us the opportunities to really use all the theory we learned in school. Some of our most challenging specimens are those with abnormal scattergrams or that have other results that just don’t make sense without further investigation and further work. Yes, they take more time but that’s what we are here for!
I work at a hospital that has a large oncology center where many sickle cell patients are treated. We also have the largest number of babies delivered in the area, and because of that there are many NICU babies. One thing these patients have in common is both populations tend to lean towards spurious reticulocyte counts with reticulocyte abnormal scattergram flags. But why?
The reticulocyte (retic) count is a good marker of erythropoietic activity of the bone marrow. Retic counts, along with the Immature Reticulocyte Fraction (IRF) are useful tools to help diagnose anemia or monitor bone marrow response to therapy. Reticulocytes are a normal stage in the development of RBCs after the nucleus has been extruded but before the cell is mature. Retics still contain RNA which is lost upon cell maturation. On automated hematology analyzers, RBCs are counted using impedance or optical technology. Automated reticulocyte counts are based on identifying young RBCs which have residual nucleic acids after expelling the nucleus. Hematology analyzers stain the RNA in young RBCS with fluorescent or non-fluorescent dyes, and principles of impedance, scatter, fluorescence and flow cytometry are used to measure the immature cells. Reticulocytes have some leftover nucleic acid content, and the youngest reticulocytes, newly out of the bone marrow have even more nucleic acid content. This causes more fluorescence intensity. On analyzers using fluorescence, reticulocytes are separated from mature RBCs by light scatter based on their fluorescence intensity. The total reticulocyte population is separated into 3 fractions of low, medium, and high fluorescence. The percentage of medium plus highly fluorescent reticulocytes represents the youngest of the young RBCs, which is the IRF. These can be visualized on the retic scattergram (figure 1).
Figure 1. Reticulocyte scattergram, Sysmex America
On an extended scattergram, signals with fluorescence intensity between reticulocytes and WBCs represent the upper particle portion (UPP) (figure 2). Interference can be seen on automated reticulocyte scattergrams when there are cells that have more nucleic acid than reticulocytes. Howell-Jolly bodies are DNA remnants seen in Wright-stained RBCs after the nucleus is extruded. These may be found in patients after splenectomy or when splenic function is compromised. They appear as single round dark blue inclusions in RBCs. In patients with Howell-Jolly bodies, interference with the reticulocyte count may be seen, triggering abnormal scattergram flags. Other peripheral blood findings that contain more nucleic acid than mature RBCs and reticulocytes may also cause interference and abnormal scattergrams in the reticulocyte channel. These include shift reticulocytes, basophilic stippling, nucleated RBCs and parasites. In patients with extreme leukocytosis, abnormal numbers of WBCS and WBC fragments may interfere with automated reticulocyte counting.
Figure 2. Extended Reticulocyte Scattergram, Sysmex America
Case 1
Newborns would be expected to have a higher percentage of reticulocytes than adults. Reference ranges for reticulocyte% in newborns is 2-6% compared to 0.5-1.5% for adults. Lower oxygen levels in the womb trigger the increased secretion of erythropoietin, which stimulates red blood cell (RBC) production. Increased production of RBCs results in a higher proportion of reticulocytes circulating in the newborn’s blood. In these cases, the bone marrow will release reticulocytes prematurely into the blood. These young reticulocytes that have more nucleic acid are called shift or stress reticulocytes. Premature infants may have even higher reticulocyte counts and more shift reticulocytes due to their accelerated development needs. As well, in babies with any form of hemolytic disease of the newborn, hemolysis is taking place, and a high reticulocyte count with shift reticulocytes is an indication that there is a compensation by producing more RBCs. In addition, oftentimes in these newborns, nucleated RBCs are present because the constant call for new RBCs leads to even more immature RBCs being released into the peripheral blood. Nucleated RBCs (nRBCs) are another interference factor in reticulocyte scattergrams.
In case 1 the newborn had an MCV of 103.3 which can be attributed to young RBCs known as reticulocytes. Reticulocytes are larger than mature RBCs because RBCs decrease in size with maturity. A high MCV and high reticulocyte(retic) count are therefore normal in newborns. A review of the smear on this baby showed nRBCs, basophilic stippling and slight polychromasia which all may interfere with automated reticulocyte counts. Diluting the samples reduced the interference and give us valid, reportable results.
Case 2
Sickle cell anemia is hemolytic anemia in which the abnormal sickle cells are being destroyed prematurely. Immature red blood cells including reticulocytes and shift reticulocytes are released prematurely from the bone marrow into the bloodstream in response to anemia, indicating the bone marrow’s attempt to compensate for a shortage of red blood cells. Sickle cell patients tend to have high reticulocyte counts, particularly during sickle cell crisis, have shift reticulocytes and often have nucleated RBCs, for the same reasons as premature newborns.
The smear on this patient showed numerous sickle cells, nRBCs, polychromasia, basophilic stippling, and Howell-Jolly bodies. In this case, however, the dilution did not eliminate the interference, and the abnormal reticulocyte flag was still present. A manual reticulocyte count using a Miller ocular was performed and the retic% was 3.2 %, quite a difference from the 12.8% and 9% from our analyzer! This illustrates the importance of resolving the discrepancy before reporting out a spurious count. After all, that’s why we have Medical Laboratory Scientists! Without our careful attention to detail, you’re just guessing.
References
Gaur M, Sehgal T. Reticulocyte count: a simple test but tricky interpretation! Pan Afr Med J. 2021 Sep 2;40:3. doi: 10.11604/pamj.2021.40.3.31316. PMID: 34650653; PMCID: PMC8490160.
Kim A, Park J, Kim M, Lim J, Oh EJ, Kim Y, Park YJ, Han K. Correction of pseudoreticulocytosis in leukocytosis samples using the Sysmex XE-2100 analyzer depends on the type and number of white blood cells. Ann Lab Med. 2012 Nov;32(6):392-8. doi: 10.3343/alm.2012.32.6.392. Epub 2012 Oct 17. PMID: 23130337; PMCID: PMC3486932.
Sysmex Customer Resource Center, Sysmex America, 2025
Uppal, V., Naseem, S., Bihana, I. et al. Reticulocyte count and its parameters: comparison of automated analyzers, flow cytometry, and manual method. J Hematopathol13, 89–96 (2020). https://doi.org/10.1007/s12308-020-00395-8
-Becky Socha, MS, MLS(ASCP)CMBBCM graduated from Merrimack College in N. Andover, Massachusetts with a BS in Medical Technology and completed her MS in Clinical Laboratory Sciences at the University of Massachusetts, Lowell. She has worked as a Medical Technologist for over 40 years and has taught as an adjunct faculty member at Merrimack College, UMass Lowell and Stevenson University for over 20 years. She has worked in all areas of the clinical laboratory, but has a special interest in Hematology and Blood Banking. She currently works at Mercy Medical Center in Baltimore, Md. When she’s not busy being a mad scientist, she can be found outside riding her bicycle.
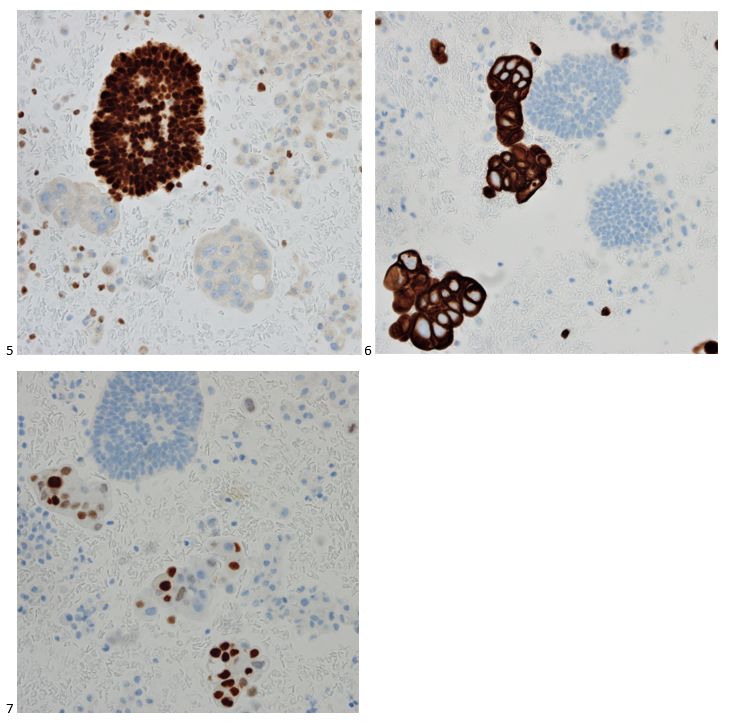
A 58-year-old woman with past medical history significant for degenerative joint disease and an approximately 30-pack per year smoking history was referred for low-dose computed tomography (CT) scan by her primary care provider to screen for lung cancer. An 8-mm nodule was observed in the lingula of the left lung. Follow up PET scans showed mild hypermetabolic activity in this nodule, as well as in axillary and subpectoral lymph nodes. The patient underwent a left video-assisted thoracic surgery to remove the nodule, and intraoperative frozen sectioning of the nodule showed necrosis but was negative for malignancy. On further discussion with the patient, it was discovered that she had spent several years living in Arizona and had participated in outdoor activities but had never observed any acute or chronic respiratory symptoms. On hematoxylin and eosin (H&E) staining of the nodule from biopsy, necrotizing granulomas and endospore-containing spherules were noted. These structures, consistent with Coccidioides, were also visualized under Gomori methenamine silver (GMS) and periodic acid-Schiff (PAS) staining, Of note, tissue culture of the specimen was negative after eight weeks (image 1).
Image 1. Lung tissue from the patient revealed spherules stained with hematoxylin and eosin (H&E) (left) and Gomori methenamine silver (GMS) (right).
Discussion
Coccidioides species, particularly C. immitis and C. posadasii, are dimorphic fungi endemic to the Western hemisphere and have been documented in North, Central, and South America. Within the United States, Coccidioides has historically been associated with the arid southwestern states, with the Centers for Disease Control and Prevention (CDC) noting that many cases of coccidioidomycosis originate in California and Arizona.1 However, both the CDC and a 2022 study evaluating rates of coccidioidomycosis diagnosis have identified cases across the country, most frequently among travelers to high-prevalence areas. In the United States, other regions of endemicity include southwestern New Mexico, Utah, Washington, and western Texas.1,2 This fungus grows best in soil as mycelia, which, in dry environments, develop into spores.3,4 These spores can be released into the air with disruption of the soil, through natural or man-made forces causing infection via inhalation. Generation of dust clouds such as through natural occurrences of earthquakes and windstorms have led to outbreaks and lead to increased risk of exposure for individuals around the area.1,2
Coccidioidomycosis, also called “valley fever”, is a syndrome characterized by pulmonary and systemic symptoms, though it is more frequently asymptomatic. The incubation period is typically 1 to 3 weeks. Patients frequently present several weeks after exposure with fever, chills, night sweats, cough, and arthralgias.4 Given the general nature of these symptoms, it is believed that many cases are undiagnosed or misattributed to other causes of community acquired pneumonia. Mild pulmonary coccidioidomycosis is frequently self-resolving over the course of weeks to months, but persisting respiratory infection in patients previously thought to have bacterial or viral pneumonia can be the first indication of a fungal etiology.3 A small percentage of affected patients can develop more complex disease, including more severe pulmonary involvement and disseminated infection. Complex pulmonary coccidioidomycosis is characterized by pleural effusion, nodules, and cavities. Empyema can occur as a consequence of fistula formation between a peripheral cavitary lesion and the pleural space.3 Disseminated disease has numerous manifestations, including cutaneous and soft tissue disease, osteomyelitis, synovitis, and meningitis.
Coccidioides can also be isolated from respiratory specimens, tissue biopsies and cerebrospinal fluid using routine fungal culture medium. Direct staining for the fungal elements (and sometimes spherules) from unfixed specimens can be done using calcofluor white, fluorescent stain. Procedures that involve manipulation of the sporulating dimorphic molds such as Coccidioides require biosafety level 2 practices and facilities and should be performed in a class II biological safety cabinet. Lifting of the culture plate lid is sufficient to release numerous spores into the environment capable of causing an infection.4 Cultured Coccidioides poses an occupational risk, as there have been reports of laboratory personnel developing symptoms of infection after working with the cultured fungus. As with other dimorphic molds, these organisms exhibit two forms: the mold form grown on Sabouraud dextrose agar or potato dextrose agar grown at 25C and a yeast form when grown at 37C. Mycelia are often visible within the first several days of incubation at 25C. The mold colonies may be white to grey, and sometimes tan or red with age. Microscopic examinations may reveal septate, hyaline, hyphae. Coccidioides form arthroconidia which are one-cell length, cylindrical to barrel-shaped with thick, smooth walls in their hyphae. The arthroconidia may alternate with empty disjunctor cells, giving the impression of alternating staining hyphae. True arthroconidia like Coccidioides will fragment. At 37C, Coccidioides develop into spherules, thick-walled structures where numerous endospores develop. The spherule form is rarely isolated in cultures but can be visualized on PAS, GMS, or H&E stains from fixed tissues.4
Diagnosis can also be made using serological testing, including enzyme immunoassay, immunodiffusion, and complement fixation IgM and IgG assays. These tests have varying sensitivities and specificities, often depending on the manufacturer of the test, though complement fixation and immunodiffusion methods in general have high sensitivities, rendering a positive result as virtually diagnostic.3 Coccidioidin is typically the antigen from the mycelial phase of the mold that is used for serological tests. A positive result from an immunodiffusion complement fixation test typically is indicative of a recent or chronic infection with IgG antibodies detected 2-6 weeks after onset of symptoms. On the other hand, the complement fixation test becomes positive 4-12 weeks after infection. Evaluating the titers can help determine the time and severity of infection. A titer <1:4 suggest early to residual disease whereas titers >1:16 could suggest disseminated disease beyond the respiratory tract.6 Any detection of antibodies from CSF is suggestive of meningitis caused by Coccidioides. Of note, in scenarios where there are discrepant results between serological tests, the patient should be investigated for histoplasmosis or blastomycosis. Antigen detection for Coccidioides species have been developed but performance is poor in urine but slightly higher in CSF specimens; similarly, Beta-D-glucan testing in serum for coccidioidomycosis also has limited value.7,8.9 Molecular methods of diagnosis have also been developed, but only one PCR test has been FDA approved with comparable sensitivity to cultures.10
Treatment of primary pulmonary and disseminated coccidioidomycosis typically consist of the triazoles such as voriconazole, posaconazole, itraconazole, and fluconazole. Amphotericin B can also be used, though this is usually reserved for poor responding or rapidly progressing infection, given its side effects.11 Immunocompromised individuals are at increased risk for disseminated infection whereas immunocompetent individuals may recover without treatment. The Infectious Diseases Society of America notes that mild cases of limited pulmonary involvement can be managed with observation and supportive measures alone.12
2. Mazi PB, Sahrmann JM, Olsen MA, et al. The Geographic Distribution of Dimorphic Mycoses in the United States for the Modern Era. Clin Infect Dis. 2023;76(7):1295-1301. doi:10.1093/cid/ciac882
3. Johnson RH, Sharma R, Kuran R, Fong I, Heidari A. Coccidioidomycosis: a review [published correction appears in J Investig Med. 2021 Dec;69(8):1486. doi: 10.1136/jim-2020-001655corr1]. J Investig Med. 2021;69(2):316-323. doi:10.1136/jim-2020-001655
4. McHardy IH, Barker B, Thompson GR 3rd. Review of Clinical and Laboratory Diagnostics for Coccidioidomycosis. J Clin Microbiol. 2023;61(5):e0158122. doi:10.1128/jcm.01581-22
5. David A. Stevens, Karl V. Clemons, Hillel B. Levine, Demosthenes Pappagianis, Ellen Jo Baron, John R. Hamilton, Stanley C. Deresinski, Nancy Johnson, Expert Opinion: What To Do When There Is Coccidioides Exposure in a Laboratory, Clinical Infectious Diseases, Volume 49, Issue 6, 15 September 2009, Pages 919–923, https://doi.org/10.1086/605441
6. Smith CE, Saito MT, Simons SA. 1956. Pattern of 39,500 serologic tests in coccidioidomycosis. JAMA 160:546–552. doi: 10.1001/jama.1956.02960420026008.
7. Durkin M, Connolly P, Kuberski T, Myers R, Kubak BM, Bruckner D, Pegues D, Wheat LJ. Diagnosis of coccidioidomycosis with use of the Coccidioides antigen enzyme immunoassay. Clin Infect Dis. 2008 Oct 15;47(8):e69-73. doi: 10.1086/592073. PMID: 18781884.
8. Kassis C, Zaidi S, Kuberski T, Moran A, Gonzalez O, Hussain S, Hartmann-Manrique C, Al-Jashaami L, Chebbo A, Myers RA, Wheat LJ. Role of Coccidioides Antigen Testing in the Cerebrospinal Fluid for the Diagnosis of Coccidioidal Meningitis. Clin Infect Dis. 2015 Nov 15;61(10):1521-6. doi: 10.1093/cid/civ585. Epub 2015 Jul 24. PMID: 26209683.
9. Thompson GR 3rd, Bays DJ, Johnson SM, Cohen SH, Pappagianis D, Finkelman MA. Serum (1->3)-β-D-glucan measurement in coccidioidomycosis. J Clin Microbiol. 2012 Sep;50(9):3060-2. doi: 10.1128/JCM.00631-12. Epub 2012 Jun 12. PMID: 22692738; PMCID: PMC3421794.
10. Vucicevic D, Blair JE, Binnicker MJ, McCullough AE, Kusne S, Vikram HR, Parish JM, Wengenack NL. The utility of Coccidioides polymerase chain reaction testing in the clinical setting. Mycopathologia. 2010 Nov;170(5):345-51. doi: 10.1007/s11046-010-9327-0. Epub 2010 Jun 10. PMID: 20535639.
11. Ampel NM. The Treatment Of Coccidioidomycosis.Rev Inst Med Trop Sao Paulo. 2015 Sep;57 Suppl 19(Suppl 19):51-6. doi: 10.1590/S0036-46652015000700010. PMID: 26465370; PMCID: PMC4711193.
12. Galgiani JN, Ampel NM, Blair JE, et al. 2016 Infectious Diseases Society of America (IDSA) Clinical Practice Guideline for the Treatment of Coccidioidomycosis. Clin Infect Dis. 2016;63(6):e112-46. doi:10.1093/cid/ciw360
–Anna Reed is currently a third year medical student at the George Washington University School of Medicine and Health Sciences. She intends to pursue a pathology residency, followed by a fellowship in forensic pathology. Her academic interests include the role of pathology in disaster scenarios, various topics in microbiology, and ways to improve autopsy practices.
-Rebecca Yee, PhD, D(ABMM), M(ASCP)CM is the Chief of Microbiology, Director of Clinical Microbiology and Molecular Microbiology Laboratory at the George Washington University Hospital. Her interests include bacteriology, antimicrobial resistance, and development of infectious disease diagnostics.
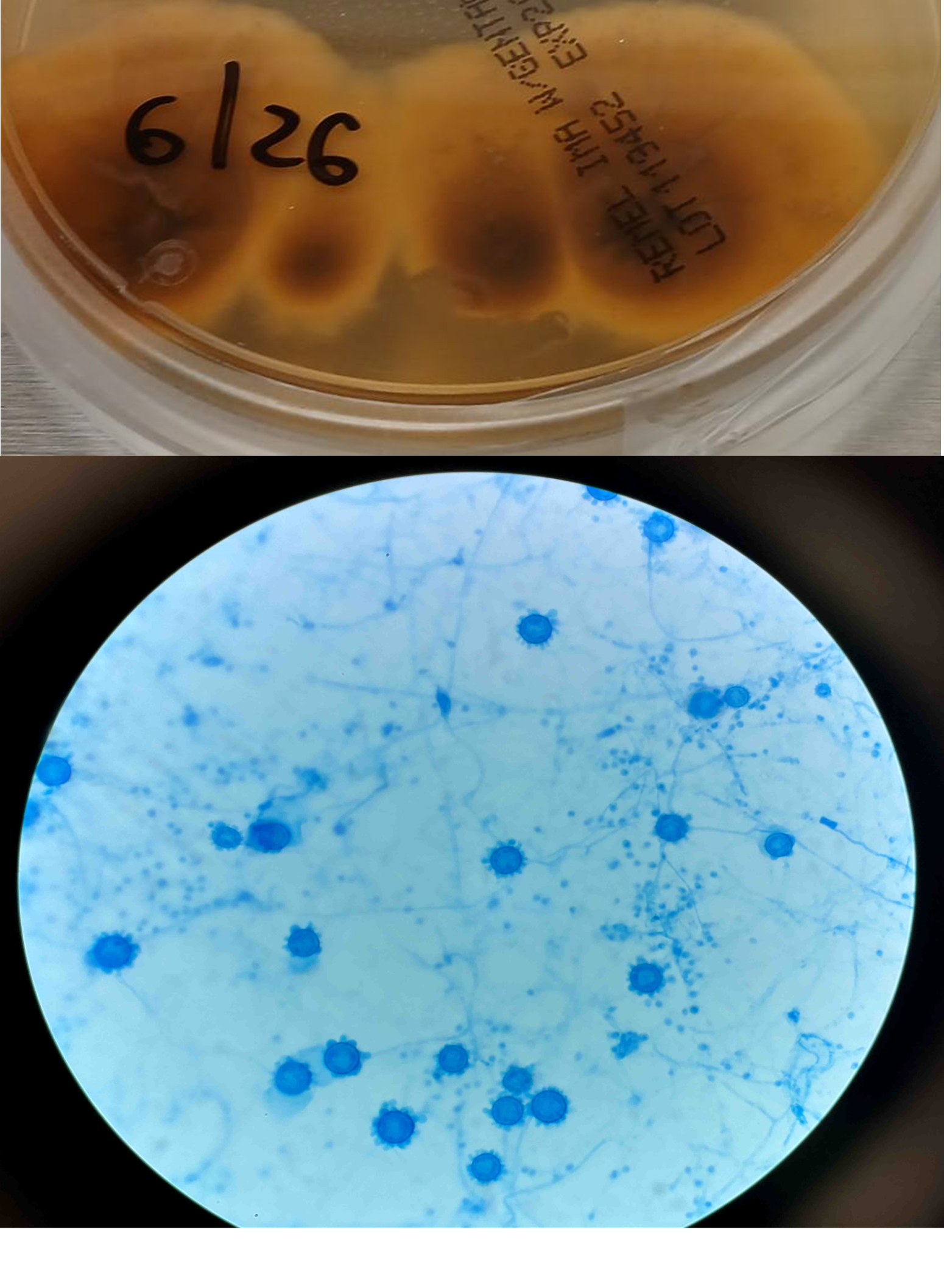
A 41-year-old male was admitted with myasthenia gravis exacerbation and respiratory difficulty. He was diagnosed with myasthenia gravis in 2004, underwent a thymectomy in 2010, and was taking prednisone (20 mg daily). He presented to another hospital one month ago with fever, chest pain, and shortness of breath. He was treated with antibiotics and antifungals for pneumonia, along with 5 days of immunoglobulin for a possible myasthenia gravis crisis, but showed no improvement. After admission, a CT scan revealed diffuse bilateral mixed airspace and ground-glass opacities without pleural effusions, most concerning for multifocal pneumonia. A bronchoalveolar lavage (BAL) with Gomori methenamine silver (GMS) staining showed hyphal and yeast elements. Both BAL and urine Histoplasma antigen tests were positive. He was started on posaconazole (300 mg daily), which led to an improvement in symptoms, and he was subsequently discharged. After 6 weeks of culture, macroscopic and microscopic features of the colony confirms Histoplasma capsulatum (Figure 1).
Figure 1. Macroscopic analysis reveals dark brown color on reverse (top). Lactophenol cotton blue preparation of mold reveals tuberculated, macroconidia (bottom).
Discussion
Histoplasma capsulatum is a dimorphic fungus responsible for histoplasmosis, a respiratory infection. As with other dimorphic molds, this fungus exists as a mold in the environment at 25°C and converts to a yeast form once it infects a host at 37°C. It is primarily endemic in areas with rich soil, particularly in the Ohio and Mississippi River valleys in the United States, but has also been reported in Maryland, Pennsylvania, Delaware, West Virginia, Virginia, and North Carolina. Outside the United States, histoplasmosis incidence is best understood and highest in parts of Mexico and South and Central America and is largely driven by the AIDS pandemic1,2. The fungus thrives in bird and bat guano due to the high nitrogen and organic content; common exposures include bird roosts, chicken houses, caves, and old buildings that are commonly visited by bats. Humans typically become infected through the inhalation of aerosolized spores. Demolition or building construction around these soiled environments can lead to outbreaks.
While most infections are asymptomatic, histoplasmosis can lead to severe illness in immunocompromised individuals. Histoplasma spores are deposited in the lungs, where they convert to the yeast form. These yeast cells are phagocytosed by alveolar macrophages, where they survive and multiply inside the cells. The fungus can then disseminate via the lymphatic and bloodstream systems to other organs 3. The clinical presentation of histoplasmosis varies depending on the host’s immune status and the level of fungal exposure. In most cases, the infection is asymptomatic or manifests as mild flu-like symptoms, including fever, fatigue, and cough. However, in cases of acute pulmonary histoplasmosis, patients may experience pneumonia-like symptoms such as chest pain, cough, and fever. Chronic pulmonary histoplasmosis can mimic tuberculosis, presenting with symptoms such as weight loss, night sweats, and a chronic cough. Pleural thickening and apical cavitary lesions are common. Extrapulmonary manifestations include granulomatous mediastinitis, fibrosing (sclerosing) mediastinitis, and broncholithiasis1. In severe disseminated cases, particularly in immunocompromised individuals, the infection can spread to multiple organs. Adrenal involvement may manifest as adrenal masses, adrenal insufficiency, or electrolyte imbalances. Gastrointestinal involvement is relatively common but rarely produces clinical symptoms4. Skin involvement occurs in up to 15% of cases in studies, more frequently in patients with AIDS. Central nervous system (CNS) involvement occurs in 5-20% of cases5.
Diagnosing histoplasmosis involves clinical suspicion, radiological studies, and laboratory tests. The most common radiographic findings are diffuse reticulonodular pulmonary infiltrates. Cavitations are seen in chronic cavitary pulmonary histoplasmosis, and mediastinal or hilar lymphadenopathy is often present. In immunocompetent individuals, infections may lead to development of granulomas that may or may not be necrotizing.
For laboratory testing, antigen detection in urine or serum using enzyme immunoassays has a sensitivity of 90%6 in disseminated disease and 75%7 in acute pulmonary disease. Antibodies may take 2-6 weeks to appear in circulation and are useful in chronic cases to confirm suspicions of infections but less effective for detecting acute infection and in immunosuppressed patients with a poor immune response.
Culturing Histoplasma from clinical specimens is definitive, but it can take 4-8 weeks due to the slow growth of the fungus. The colonies appear white at young growth, have a cottony, cobweblike-aerial mycelium and can mature into brown or grey color on reverse (Figure 1, top) 8. Microscopic examination of mold colony using lactophenol cotton blue preparations reveals the characteristic large, rounded, tuberculate macroconidia (circular structures with roughened/spiked edges) originating from short, hyaline conidiophores (Figure 1, bottom). Histoplasma capsulatum appears as small (2-5 μm), oval, intracellular yeast cells typically found within macrophages. The yeast may exhibit a clear space which may appear like a capsule, but is actually a retraction artifact due to the processing of the specimen. Staining with GMS or periodic acid-Schiff (PAS) highlights the yeasts9. Molecular methods such as PCR and sequencing have been shown to detect cases of Histoplasma as well.
Treatment varies depending on the severity of disease. Mild cases, particularly those involving acute pulmonary histoplasmosis, often resolve without specific treatment, although antifungal therapy (e.g., itraconazole) is recommended in more severe cases. For chronic or disseminated histoplasmosis, initial treatment with amphotericin B is often followed by long-term itraconazole therapy. The prognosis is favorable for immunocompetent individuals with mild disease, but disseminated histoplasmosis can be fatal in immunocompromised patients if not treated promptly.
References
1. Wheat LJ, Azar MM, Bahr NC, Spec A, Relich RF, Hage C. Histoplasmosis. Infect Dis Clin North Am. 2016 Mar;30(1):207-27. doi: 10.1016/j.idc.2015.10.009. PMID: 26897068.
2. Bahr NC, Antinori S, Wheat LJ, Sarosi GA. Histoplasmosis infections worldwide: thinking outside of the Ohio River valley. Curr Trop Med Rep. 2015 Jun 1;2(2):70-80. doi: 10.1007/s40475-015-0044-0. PMID: 26279969; PMCID: PMC4535725.
4. Sarosi GA, Voth DW, Dahl BA, Doto IL, Tosh FE. Disseminated histoplasmosis: results of long-term follow-up. A center for disease control cooperative mycoses study. Ann Intern Med. 1971 Oct;75(4):511-6. doi: 10.7326/0003-4819-75-4-511. PMID: 5094067.
5. Wheat LJ, Batteiger BE, Sathapatayavongs B. Histoplasma capsulatum infections of the central nervous system. A clinical review. Medicine (Baltimore). 1990 Jul;69(4):244-60. doi: 10.1097/00005792-199007000-00006. PMID: 2197524.
6. Wheat LJ, Kauffman CA. Histoplasmosis. Infect Dis Clin North Am. 2003 Mar;17(1):1-19, vii. doi: 10.1016/s0891-5520(02)00039-9. PMID: 12751258.
8. Azar MM, Hage CA. Laboratory Diagnostics for Histoplasmosis. J Clin Microbiol. 2017 Jun;55(6):1612-1620. doi: 10.1128/JCM.02430-16. Epub 2017 Mar 8. PMID: 28275076; PMCID: PMC5442517.
9. Hage, C. A., Ribes, J. A., Wengenack, N. L., Baddour, L. M., Assi, M., McKinsey, D. S., … & Wheat, L. J. (2010). A multicenter evaluation of tests for diagnosis of histoplasmosis. Clinical Infectious Diseases, 50(4), 508-512.
-Xingbang Zheng, M.D., was born and raised in Hefei, China. He attended the Peking University Health Science Center, where he received his doctorate degree. He then worked as an OB/GYN physician in China, primarily focusing on female infertility and reproductive surgery. His clinical research concentrated on subtle distal fallopian tube abnormalities and their relationship with endometriosis and infertility. In his free time, Xingbang enjoys swimming, visiting museums, and spending time with his family. Xingbang is currently pursuing AP/CP training.
-Rebecca Yee, PhD, D(ABMM), M(ASCP)CM is the Chief of Microbiology, Director of Clinical Microbiology and Molecular Microbiology Laboratory at the George Washington University Hospital. Her interests include bacteriology, antimicrobial resistance, and development of infectious disease diagnostics.
A 67 year old female presented to the emergency department with worsening chest pain and shortness of breath for several weeks. Her medical history was notable for diffuse large B cell lymphoma, for which she had started treatment with rituximab and tafastimab-cxix. Her complete blood count revealed severe leukopenia. A chest computed tomography scan showed focal consolidation in the right lower lobe, and broad-spectrum antibiotics and Filgrastim were started to treat her pneumonia. However, her symptoms did not improve. She underwent a bronchoalveolar lavage, which was sent to microbiology for culture. The initial Gram stain of the specimen was unrevealing. Twelve days later, a pathogen was isolated from cultures with acid-fast media and fungal media. On a Sheep’s blood agar plate, white, chalky colonies appeared. A Gram stain of the isolate showed gram-positive organisms growing as branching, beaded filaments. The organisms were further highlighted on a partial acid-fast stain. MALDI-TOF identified the organism as Nocardia farcinica. The patient was started on trimethoprim/sulfamethoxazole and imipenem, and was discharged following clinical improvement.
Images of Sheep’s blood agar plate showing white, dry colonies (left) and Gram stain highlighting beaded, branching filamentous bacteria after culture in MGIT broth (right).
Discussion
Once mistakenly classified as a fungus due to its filamentous, branching morphology, Nocardia spp. are actually gram-positive, aerobic bacteria that belong to the order Cornybacteriales1. Nocardia are normal inhabitants of the soil, where they digest decaying plant matter. However, they are a cause of human disease in susceptible hosts when they are inhaled or enter via the skin2,3 . The major risk factor for infection is an immunocompromised state, particularly defects in cell-mediated immunity. As such, patients with AIDS or lymphoma, those receiving allogeneic organ transplants, and those taking immunosuppressive drugs including high-dose steroids either are more susceptible to infection or suffer more severe disease2,4. Among persons with intact immune defenses, underlying lung diseases such as chronic obstructive pulmonary disease or cystic fibrosis, predispose to infection1.
The most common clinical manifestation of Nocardiosis, particularly in immunocompromised hosts, is a subacute to chronic pneumonia that can wax and wane, and abscesses are a characteristic pathologic feature4. Nocardia can spread via the blood to affect virtually all other organs, with the brain, eyes, and joints being common sites of dissemination1,5,6. Skin infections, more common in immunocompetent persons, include cellulitis and a lymphocutaneous disease that mimics Sporotrichosis4,5. Specifically for Nocardia farcinica, this species have been shown to be involved in disseminated diseases, with most patients being immunocompromised, but in immunocompetent individuals, cutaneous infections have been reported.
Often the first diagnostic clue comes from visualization of gram-positive beaded, thin, branching organisms on direct smears of specimens7. Of note, while the beaded, branching morphology is characteristic, this appearance is sensitive to several culture conditions including media and temperature. Given that Nocardia does not stain fully on Gram stain, it is recommended that a modified acid-fast stain be done as a reflex stain to confirm the suspicions. Organisms, aside from true Mycobacteria, that contain mycolic acids on the cell wall include Nocardia, Gordonia, Dietzia, Rhodococcus, Segmiliparus, Tsukamurella, and Williamsia. However, in some cases, not all organisms can be seen on the direct smear, which warrants cultures to rule out infection but Nocardia spp. can be difficult to isolate in culture either due to low numbers in the initial specimen, or overgrowth by contaminating bacteria3. They grow slowly, usually over the course of weeks3,4. Nocardia can be isolated from most media used to culture bacteria, fungus and mycobacteria3,7. Colonies often appear white and dry but appearance of velvety, powdery, wrinkled, or being heaped are also not uncommon. On the reverse of the plate, the colors may vary and can be brown, tan, pink, orange, red, purple, gray, yellow, peach, or white. The partial acid-fastness of Nocardia spp. is an important corroborating piece of evidence7. Molecular and proteomic methods, including 16S rRNA sequencing and MALDI-TOF allow for definitive confirmation1,7.
A diagnosis of Nocardiosis aids in the selection of appropriate antibiotics and may raise suspicion for disseminated disease, such as brain abscesses. Trimethoprim/sulfamethoxazole remains the antibiotic of choice, and patients usually respond within weeks4. Adding another potent antibiotic such as imipenem helps treat more severe or disseminated disease6. Studies done in-vitro have shown that isolates with resistance to Trimethoprim/sulfamethoxazole are typically susceptible to carbapenems.
References
1. Traxler RM, Bell ME, Lasker B, Headd B, Shieh WJ, McQuiston JR. Updated review on Nocardia species: 2006-2021. Clin Microbiol Rev. 2022;35(4).
2. Steinbrink J, Leavens J, Kauffman CA, Miceli MH. Manifestations and outcomes of nocardia infections. Medicine (Baltimore). 2018;97(40).
-Stevephen Hung MD, PhD is currently a PGY-4 resident at George Washington University Hospital. He graduated from Case Western Reserve University School of Medicine in Cleveland, OH. His academic interests include transfusion medicine, informatics and molecular pathology.
-Rebecca Yee, PhD, D(ABMM), M(ASCP)CM is the Chief of Microbiology, Director of Clinical Microbiology and Molecular Microbiology Laboratory at the George Washington University Hospital. Her interests include bacteriology, antimicrobial resistance, and development of infectious disease diagnostics.
Disseminated intravascular coagulation (DIC) is a serious process in which the proteins responsible for clot formation become overactive. It is vital to note that DIC is not a disease in itself, rather it is always secondary to an underlying disorder, which causes over-activation of the coagulation system.1 This over-activation results in activation of coagulation that consumes platelets, clotting factors, and causes microvascular fibrin thrombi, which can lead to organ failure secondary to tissue ischemia.2 The brain, kidneys, liver, and lungs are the most prone to tissue damage in DIC.
Some symptoms of DIC may include bleeding (from multiple sites), blood clots, bruising, hypotension, shortness of breath, fever, confusion, memory loss, or behavior change.3 Patients always present with symptoms of other disorders frequently associated with DIC such as sepsis, trauma, liver disease, obstetric complications, and malignancy to name a few. In most cases, coagulation does not result in clinical complications, and is not evident until the consumption of platelets and factors becomes overwhelming.1 Once there is overwhelming consumption, it becomes visible through prolonged clotting tests and increasing thrombocytopenia.1 Therefore, timely recognition is crucial in recognizing DIC.
A major challenge of both prognosis and management, of DIC is that it is not a single disease entity. Prognosis largely depends on the treatment of the underlying condition, as the condition is what drives the activation of the coagulation cascade. Treatment is generally supportive, and entails supporting various organs. Support may include the use of ventilators, hemodynamic support, or transfusion of blood components. As well, algorithms have been developed to help guide the management of patients with DIC.
Case presentation
A 31-year-old female presented to the emergency department with complaints of bleeding gums, bruising all over her body, and recurrent nosebleeds for longer than a week. The patient has a history of Acute Promyelocytic Leukemia (APL/AML-M3), and has had symptoms of gingival bleeding, widespread ecchymosis, and recurrent epistaxis for longer than a week. Past fluorescence in situ hybridization (FISH) of the patient’s bone marrow aspirate revealed a t(15;17) translocation. Bone marrow studies also showed blast cells with granulation, dysmyelopoiesis, and 60% promyelocytes with the presence of Auer rods. Immunophenotyping was negative for expression of CD34, and positive for expression of CD13 and CD33. Cytochemical staining tests were positive with myeloperoxidase (MPO), Sudan Black B (SBB), and specific esterase (SE), and negative with non-specific esterase (NSE). As well, reverse transcriptase-polymerase chain reaction (PCR) confirmed the PML-RAR(alpha) fusion gene, consistent with a diagnosis of AML-M3.
The patient has undergone one past treatment of retinoic acid, as well as traditional chemotherapy. It should be noted that some studies indicate a higher incidence of DIC in acute leukemia patients during remission induction with chemotherapy.1 The patient has no family history of the disorder/condition. There are no occupational or social implications/factors, but it is associated with a specific genetic mutation. The patient has no known drug allergies.
Vital signs were normal upon arrival. Upon physical examination, the patient appeared pale and lethargic, displayed widespread ecchymosis, and swollen gums.
Laboratory tests revealed a decreased hemoglobin concentration of 6.0 g/dL (normal, 12.0-16.0 g/dL), and an increased white blood cell (WBC) count of 19.5 x 109/L (normal, 4.5-11.0 x 109/L). The WBC differential showed 91% promyelocytes with heavy granulation, 3% myelocytes, 4% metamyelocytes, and 2% neutrophils. Platelet count was decreased at 72,000/uL (normal, 150,000-450,000/uL). Prothrombin time (PT) was prolonged at 22 seconds (normal, 10-13 seconds) and activated partial thromboplastin time (aPTT) was prolonged at 54 seconds (normal, 23-36 seconds). D-dimer concentration showed an increase at 12 ug/mL (normal, <0.5 ug/mL), and a decreased fibrinogen concentration of 60 mg/dL (normal, 200-400 mg/dL). Plasma levels of plasminogen were 65% (normal, 80-120%), and plasma levels for a2-antiplasmin were 46% (normal, 80-100%). These findings are consistent with DIC with marked hyperfibrinolysis, as correlated by low levels of a2-antiplasmin and plasminogen. The patient was diagnosed as having symptoms associated with DIC secondary to AML-M3.
The treatment for this patient included avoidance of all invasive procedures including biopsies or intravenous (IV) line placement, as the patient was at high risk for bleeding. Likewise, due to the high risk of bleeding, the use of heparin as a supportive therapy was not advocated. The patient received a platelet transfusion, with an aim of a platelet count greater than 50 x 109/L. As well, the patient received fresh frozen plasma (FFP) and fibrinogen concentration, guided by the concentration in the patient’s plasma. Supportive treatment will continue to be maintained during remission induction of AML-M3, and the disappearance of the coagulopathy.
Discussion
The typical symptoms of DIC are generally those of the underlying process, which incites the condition. Underlying conditions may include, but are not limited to, severe infection with any microorganism, sepsis, trauma, solid tumors and malignancies, hepatic failure, and obstetric complications.4 Acute DIC may present as ecchymosis or petechiae. Additionally, there is usually a patient history of bleeding such as gastrointestinal or gingival bleeding.4 Post-operative DIC patients can also experience bleeding from various surgical sites, or within serous cavities.5 One should remain cognizant of signs and symptoms of deep vein thrombosis (DVT), as well as microvascular thrombosis. One hallmark of DIC is bleeding from at least three unrelated sites.4 Chronic DIC may present as thrombosis due to excess thrombin formation. Patients with comorbid liver disease may also present with jaundice, while patients with pulmonary involvement may present with a cough, dyspnea, and hemoptysis.4
Epidemiology
DIC occurs in all ages and races, and has not been shown to have a predisposition for a particular gender.4 DIC has been known to develop in about 1% of all hospitalized patients.4 DIC accounts for 9%-19% of intensive care unit (ICU) admissions.2 Although assigning values to DIC-related morbidity and mortality proves difficult, studies show it occurs in roughly 35% of septic patients.1 In a trauma setting, the presence of DIC may often double the mortality rate.4 DIC may be diagnosed in roughly 15% of patients with underlying acute leukemia.1 Previous studies report the occurrence of major bleeding to be 5%-12%, although patients with a platelet count of less than 50 x 109/L show a higher risk for bleeding when compared to patients with increased platelet counts.1 The occurrence of thrombosis in both small and midsize vessels is more commonly seen, which contributes to organ failure. Overall, the severity of DIC is directly related to increased mortality, thus emphasizing the importance of early recognition.
Related Disorders/Conditions
Several conditions are known to be complications of DIC. These conditions include, but are not limited to, kidney injury, altered mental status, respiratory dysfunction, hemothorax, gangrene, digit loss, and shock.4 In most cases, the release of tissue material into circulation in conjunction with endothelial disruption, leads to the activation of the coagulation cascade.
Pathophysiology
In DIC, the patient’s underlying condition stimulates powerful procoagulant activity that results in excess thrombin. The excess thrombin then overcomes the anticoagulant control mechanisms of protein C (PrC), antithrombin (AT), and tissue factor pathway inhibitor (TFPI), allowing for widespread thrombosis throughout the vasculature.2 The patient paradoxically battles between an excess thrombin state due to thrombin, embolism, and microvascular occlusion, and a hemorrhagic disorder secondary to depletion of platelets, consumption of coagulation factors, and accelerated formation of plasmin.2
To fully understand DIC, one must review normal hemostasis. Normal hemostasis generally takes place in response to an injury to the blood vessel wall. In normal hemostasis, a well-maintained process takes place in three stages: primary hemostasis, secondary hemostasis, and fibrinolysis. Primary hemostasis results in formation of the primary platelet plug via platelet adhesion, aggregation, and secretion.6 Secondary hemostasis is responsible for the formation of fibrin, and involves plasma proteins. In secondary hemostasis, fibrinogen is converted to fibrin, which forms a mesh that acts to reinforce the primary platelet plug, thus forming a hard, more stabilized clot at the site of injury, stopping any active bleeding.6 Finally, fibrinolysis takes place. Fibrinolysis is the process of the removal of fibrin through proteolysis, so that normal vessel structure and function can be restored.6 As part of normal healing, in response to an injury, clotting should only occur as a localized phenomenon, as there are many checks and balances to prevent extension of the hemostatic plug mechanism to the entire intravascular system.2 In DIC, this process of hemostasis runs out of control.
Laboratory Tests and Applications
Since DIC is secondary to an underlying disease, its diagnosis can be challenging, as there is no single laboratory test with sufficient accuracy to allow for a diagnosis. Imaging studies are generally only useful in detecting the underlying condition inciting DIC.4 DIC may present with a wide range of abnormalities in laboratory values. Laboratory findings of thrombocytopenia, prolonged coagulation times, decreased fibrinogen, elevated fibrin degradation products (FDPs), and elevated D-dimer levels are indicative of DIC.4 A complete blood count (CBC) and peripheral blood smear may show moderate-to-severe thrombocytopenia, as well as the presence of schistocytes secondary to evidence of microangiopathic pathology.4 Additionally, scoring systems have been developed to aid in diagnosis of DIC.4 The International Society on Thrombosis and Hemostasis (ISTH) developed a scoring system that makes use of laboratory tests, and a DIC score calculator. However, it is important to note, the presence of an underlying condition frequently associated with DIC is essential for the use of this diagnostic algorithm.4 Studies have reported the sensitivity of the DIC as 93%, and the specificity as 98%.4 As well, this scoring system has shown to be a strong predictor for mortality in sepsis. Similar scoring systems have been developed in other countries as well. Other conditions that may be ruled out by these tests include thrombotic thrombocytopenic purpura (TTP), hemolytic uremic syndrome (HUS), immune thrombocytopenia (ITP), chronic liver disease, Heparin-induced thrombocytopenia (HIT), and Coumadin/Vitamin K deficiency.2
Correlation of Laboratory Tests
Thrombocytopenia is the most common laboratory finding in DIC, reported in 93% of cases.5 In many cases, the thrombocytopenia may not yet be severe; therefore, it is crucial to recognize a downward trend in platelet count despite a recent count in a normal range.5 Acute DIC typically presents with a rapid consumption of platelets and clotting factors, so that at the time of diagnosis, the platelet count may be less than 50 x 109/L, the PT and aPTT values are markedly prolonged.2 FDPs appear elevated, as they are released into circulation as the body attempts to break down clots. Fibrinogen levels may be decreased, as it is used up. Likewise, plasminogen and AT show a decrease.2 D-dimers appear increased as a result of fibrinolysis and excess plasmin.2 Red blood cells (RBCs) that are pushed through occluded vessels in the microvasculature become fragmented and will often appear on a peripheral blood smear as schistocytes.
Impact of Pathophysiology on Laboratory Results
DIC can present with a variable range of abnormal laboratory values. Additionally, the values may only represent a momentary glimpse into a rapidly changing systemic process and often requires frequent repeated testing.4 As well, patients with chronic DIC may present with only minimal abnormalities in laboratory tests. There is no single test that is a specific indicator for DIC. Rather, the use of combined tests such as platelet count, PT, aPTT, fibrinogen, and D-dimer, in conjunction with an underlying disorder, help guide management and treatment.
A diagnosis of DIC is reached only through a combination of the clinical impression in conjunction with laboratory abnormalities.5 The patient history plays a significant role in that DIC is always secondary to an underlying condition, therefore the clinical presentation of DIC will be the features of the primary disease.
Treatment
The treatment of DIC typically involves focusing on the underlying disorder, as manifestations of DIC will subside once the underlying condition is addressed. Management of DIC itself follows four basic features: monitoring vital signs, assessing and documenting the extent of thrombosis and hemorrhage, correcting hypovolemia, and administering basic hemostatic procedures when indicated.4 Therapeutic interventions are mostly supportive in nature, and may include transfusion of blood components including platelets and clotting factors. However, blood components should be reserved for those patients who have hemorrhage, require a surgical procedure, or pose a high risk for bleeding complications.2 The use of heparin is usually reserved for cases of chronic DIC, and should be provided to patients demonstrating widespread fibrin deposition without evidence of substantial hemorrhage.4 Generally, the use of antifibrinolytic drugs should be avoided, as they have been known to cause thrombotic complications, although these agents have proven useful in cases associated with AML-M3 and other types of cancer.4 In these cases, Heparin should always be administered with antifibrinolytic drugs so as to arrest any prothrombotic effects.4 In patients with trauma and massive blood loss, antifibrinolytics such as tranexamic acid has shown to be effective in reducing blood loss and improving survival.4 Only minimal cases have been reported to benefit from administration of activated PrC.
Patient Response to Treatment
Treatment decisions for this patient are based on clinical and laboratory evaluation of hemostasis. The patient is to avoid all invasive procedures such as IV-line placement and biopsies. Heparin, in this case, is contraindicated secondary to bleeding risk. The patient received FFP, platelets, and fibrinogen concentrate. The patient currently remains hemodynamically stable, however, if evidence of significant bleeding and hyperfibrinolysis remains, tranexamic acid may be administered in conjunction with heparin. Clinical and laboratory parameters will continue to be monitored every eight hours.
Prognosis
The prognosis of patients with DIC depends on the severity of the coagulopathy, as well as the underlying condition that incited DIC. Generally, if the underlying condition is self-limiting or can be appropriately managed, DIC will disappear, and coagulation status will return to normal.4 Since DIC is a complex process, it is crucial to monitor the patient for clinical improvement or worsening, as well as to identify the early development of possible complications.
Treating the underlying AML-M3 is critical for the prognosis and management of this patient. The patient is encouraged to regularly consult with a hematologist for assistance with management of DIC, as chronic DIC may be treated on an outpatient basis after stabilization.4 Chronic DIC in patients with cancer may be managed with low molecular weight heparin or subcutaneous heparin. As well, other subspecialty consultations are necessary as indicated by the patient’s primary diagnosis.
Conclusion
DIC is not a specific illness, rather, it is a complication or an effect secondary to the progression of other underlying illnesses. It is associated with numerous clinical conditions, which involve excessive activation of the coagulation system.
Although interventions are supportive, they have been shown to be only partly effective. Since the clinical course of DIC ranges from asymptomatic to life-threatening, understanding its pathogenesis is vital for early recognition. Despite diagnostic criteria to aid physicians in the management of patients with DIC, newer laboratory markers and treatment modalities are warranted to improve patient outcome.
References
1 Levi M, Scully M. How I treat disseminated intravascular coagulation. Blood. 2018 Feb 22;131(8):845-54. doi: 10.1182/blood-2017-10-804096
2 Boral BM, Williams DJ, Boral LI. Disseminated intravascular coagulation. Am J of Clin Pathol. 2016 Dec;146(6):670-68. doi: 10.93/AJCP/AQW195
3 Medline Plus [Internet]. Bethesda (MD): National Library of Medicine (US); updated [date]. Disseminated intravascular coagulation; [updated 2022 Mar 21; reviewed 2019 Sep 24; cited 2022 Mar 30]; [about 2 p.] Available from: https://medlineplus.gov/ency/article/000573.htm
5 Thachill J. Disseminated intravascular coagulation: A practical approach. Anesthesiology. 2016 Jul;125(1): 230-6. doi: 10.1097/ALN.0000000000001123
6 Rodak BF, Fritsma MG, Fritsma GA. Normal hemostasis and coagulation. In: Hematology: Clinical principles and applications. St. Louis, MO: Elsevier Saunders; 2012. p. 626–44
7 Hussain SA, Zafar A, Faisal H, Vasylyeva O, Imran F. Adenovirus-associated disseminated intravascular coagulation. Cureus. 2021 Mar 30;13(3)e14194. doi: 10.7759/cureus.14194 8 Onishi T, Ishihara T, Nogami K. Coagulation and fibrinolysis balance in disseminated intravascular coagulation. Pediatr Int. 2021 Nov;63(11):1311-18. doi: 10.1111/ped.14684
-Kaysi Bujniewicz, MLS(ASCP)CM graduated Magna Cum Laude from University of North Dakota School of Medicine with a Bachelor of Science in Medical Laboratory Science. She has worked in clinical laboratories for over eight years as a certified and licensed Medical Laboratory Technician and a Medical Laboratory Scientist. Although her true callings are in Immunohematology and Clinical Microbiology, she currently works as a Generalist.
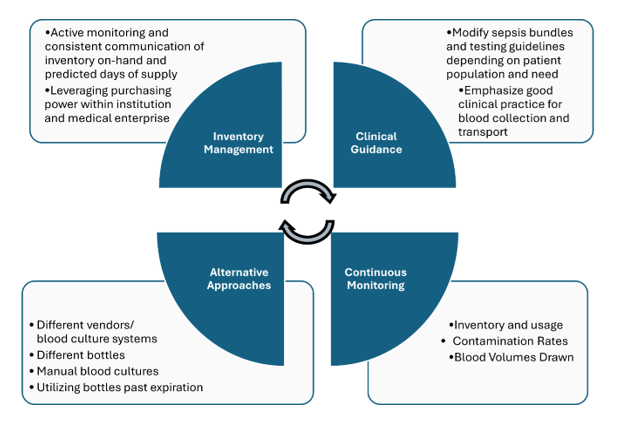
In June 2024, BD (Becton Dickinson) notified their customers that there will be a global shortage of BACTEC blood culture bottles which is anticipated to last several months. Blood culture bottles are critical for diagnosing infections in patients, and their scarcity could lead to delays in diagnosis and challenges in managing patient care, especially for those with serious systemic infections. During supply shortages, it is paramount that management relies heavily on careful inventory oversight, and prudent use and stewardship of available reagents. Here we will summarize key measures recommended to ensure continuity of care. However, it must be emphasized that there is no ‘one size fits all’ approach and each institution need to consider what works best for their patients and workflow. It is a team effort-laboratorians must work collaboratively with infectious disease providers, hospital leadership, supply management teams and other stakeholders to implement and manage efforts without duplication of efforts.
Figure 1. Summary of key measures used in dealing with blood culture bottle supply shortage
Inventory Management:
There needs to be continuous monitoring and communication among all stakeholders regarding inventory and usage. It is imperative that hospital leadership are aware of the current inventory on-hand and the predicted days of supply.
If the hospital is within a medical center, consider centralizing supplies and reallocating to those in dire need. If your institution is part of a larger system or corporation, consider leveraging system’s purchasing power vs. a single institution purchasing power to ensure availability of the blood culture bottles.
Clinical guidance:
In many institutions, laboratory and infectious disease teams, in collaboration with the clinical partners, developed sepsis bundles, in which blood cultures and lactic acid testing are the corner stones of the bundle. Most of the time, such bundles are built around the CMS guidelines, as part of the overarching Medicare’s Hospital Value-Based Purchasing Program (VBP). Such bundles are great tools to promote the best standard of care practices, as well as serving as a standard communication tool. Most organizations institute two separate types of sepsis bundles: the severe sepsis bundle and the sepsis shock bundle. With time, we need to consistently leverage quality and patient safety approaches, as well as regulatory standards to ensure the most optimal test utilization overall, especially during the times of shortages. Implementation of organization testing guidelines in the bundles promotes standardization and compliance.
Before ordering any tests for infectious diseases, it is crucial to carefully evaluate the pre-test probability of bacteremia and the likelihood of identifying a clinically significant organism. Repeating blood cultures within 48 hours of the initial set typically yields low results. Blood cultures should be collected before initiation of antimicrobial therapy. Indications for blood culture draws may vary depending on the patient population (e.g. adults versus pediatrics, immunocompetent versus neutropenic, inpatients versus from the emergency department) [1, 2]. For certain patient populations, consider performing only two blood cultures (1 set) vs. widely accepted and recommended four blood cultures (2 sets). While this is beyond the scope of this blog post, please refer to the sources below for specific clinical guidance.
Due to the typically low levels of bacteria present in the bloodstream, collecting a larger volume of blood increases the chances of obtaining a positive culture. To maximize the likelihood of detecting bacteria, blood culture bottles should be filled with the optimal volume recommended by the manufacturer, typically 8-10 mLs up to the fill line, and be sent to the microbiology laboratory in the timely manner. Reducing contamination in blood cultures decreases the need for additional resources, such as repeat blood cultures, unnecessary antibiotic treatments, and further diagnostic tests. Contamination can be minimized by strictly adhering to antiseptic procedures at collection sites, ensuring proper training for collectors. It is also crucial to remind healthcare providers and clinical staff to follow institutional guidelines for proper specimen labeling, maintaining specimen stability, and adhering to transport instructions to reduce the risk of sample rejection and prevent unnecessary waste of supplies [3].
Thinking outside the box:
Despite the shortage, patient care continues, and laboratories may need to think of alternative approaches. As of writing this post, other blood culture systems and their respective bottles are not affected. That said, implementing new instruments and switching to an alternative supply chain from a different manufacturer may be an option but often can take up to several months due to contractual requirements. Additionally, massive re-education and training (laboratory staff, phlebotomy, and nursing) will need to take place as different systems have unique collection procedures. Some institutions may opt to send out their blood cultures to reference laboratories that use an alternative system, although turnaround time may be an issue. Manual blood cultures, although laborious, could be set up and be done in-house. While it is not recommended to use expired blood culture bottles, recent published studies have shown that expired bottles up to 3 months are just as effective [4, 5]; institutions should maintain expired bottles if regulatory bodies allow for their utility at an later date as BD have extended the expiration date for certain lots already (see BD manufacturer webpage below). BD has recently announced the re-introduction of anaerobic media in glass bottles. Laboratories should prepare staff and develop plans for validation work to ensure that these bottles can be rapidly implemented for usage.
Continuous monitoring:
Aside from monitoring inventory and bottle usage, it is important for laboratories to regularly monitor contamination rates and implement a quality system that tracks blood culture volumes. To allow for targeted and more efficient education, monitoring trends stratified by different departments can help offer specific feedback to the clinical partners collecting the samples.
As we implement additional testing methodologies, remember to continuously revise your Clinical Practice Guidelines (CPGs), order sets, test utilization, etc. as part of such implementations.
A lesson learned from the COVID-19 pandemic is the need to be agile and quickly adapt to the changes in supply chain. For many laboratories, this was an effective strategy to ensure testing availability for all patient populations. Although this blood culture bottle shortage is a crisis many are facing, implementing diagnostic stewardship for blood culture collection may have overall benefits such as decreasing the amount of unnecessary testing, treatment of clinically insignificant infections, unnecessary blood draws, and potential central line-associated bloodstream infections.
Here are external resources/references available to help laboratories and healthcare providers:
Manufacturer’s web page: https://bdbactec-update.com/
American Society for Microbiology (ASM) guideline, endorsed by the Society for Healthcare Epidemiology of America (SHEA): https://asm.org/guideline/blood-culture-shortages-management-diagnostic-stew
Informational web page from the Infectious Disease Society of America (IDSA): https://www.idsociety.org/clinical-practice/blood-culture-bottle-shortage/
References
1. Fabre, V., K.C. Carroll, and S.E. Cosgrove, Blood Culture Utilization in the Hospital Setting: a Call for Diagnostic Stewardship. J Clin Microbiol, 2022. 60(3): p. e0100521.
2. Fabre, V., et al., Principles of diagnostic stewardship: A practical guide from the Society for Healthcare Epidemiology of America Diagnostic Stewardship Task Force. Infect Control Hosp Epidemiol, 2023. 44(2): p. 178-185.
3. Clinical and Laboratory Standards Institute, Principles and Procedures for Blood Cultures, CLSI guideline M47. 2nd Edition. 2022.
4. Hardy, L., et al., Blood culture bottles remain efficient months after their expiration date: implications for low- and middle-income countries. Clin Microbiol Infect, 2024. 30(10): p. 1327-1328.
5. Klontz, E.H., et al., Evaluation of expired BD BACTEC blood culture vials. J Clin Microbiol, 2024: p. e0108224.
-Rebecca Yee, PhD, D(ABMM), M(ASCP)CM is the Chief of Microbiology, Director of Clinical Microbiology and Molecular Microbiology Laboratory at the George Washington University Hospital. Her interests include bacteriology, antimicrobial resistance, and development of infectious disease diagnostics.
-Olga Kochar, MS, CSSGB, is the Divisional Director of Laboratory and Transfusion Services with the GW Hospital, as well as faculty with the GWU School of Medicine and Health Sciences. With over 29 years in the healthcare industry, Olga Kochar is passionate about quality and patient safety, performance improvement and teaching.
Back in the olden days – wait, can I use that line yet? I graduated from my cytology program ten years ago, which feels like two decades ago or just yesterday. Depends on the day. Anyway, there are little nuggets from my training that stick with me whenever I’m screening a case. This case revolves around the concept of a two-cell population in body fluids, namely pleural, peritoneal, and pericardial. We were taught that if you see a two-cell population, the case is malignant. I remember asking in school, “But what if the diagnosis is mesothelioma?” “Okay,” my professor said, “that applies to any malignancy except mesothelioma. The two-cell population indicates a population of benign mesothelial cells and a second population of malignant cells.” Noted! I live by this rule, and every now and then, I have a case of a peritoneal fluid where the slides are covered in “wall-to-wall” adenocarcinoma, and I can’t find a benign mesothelial cell for the life of me. Obviously, we let years of experience with morphology take the reins there, but there are still those cases where the native mesothelial cells are so reactive in appearance that we start hunting for a two-cell population. This case was a perfect blend of wall-to-wall tumor and more-than-reactive mesothelials.
When a patient is being worked up for the first time or the nth time, we recognize their names and recall their clinical history and previous morphologies. Quite often we have patients with genetic predispositions to cancer who present with one type of cancer and are treated accordingly, and during surveillance imaging, come back to us with a second primary cancer. In this case, we received a right-sided pleural fluid on a 73-year-old woman with a history of ER+/PR+/HER2- breast cancer. After a history of incomplete medical follow-up, the patient presented to a local emergency room with pleuritic pain, upon which a CT Scan identified a lung mass, breast mass, and multiple liver and upper thoracic bone lesions. The assumption, given the clinical history, was metastatic breast cancer, and the clinician submitted the pleural fluid for cytology to repeat ER/PR/HER2 testing to determine if there is a better treatment strategy than her current aromatase inhibitor, which appeared to be failing.
Upon screening the cytopreparations, we observed very obvious malignant cells (Image 1). It was evident that we had a two-cell population. One of the cell populations was characterized by cohesive clusters of relatively uniform tumor cells (Image 2 & 4) and the second cell population was less cohesive with larger pleomorphic cells (Image 3 & 4). Hmm, something doesn’t make sense here. We already have a group of tumor; the other cells are supposed to be benign mesothelials cells. Is there a mixed morphology going on? Like a combined small cell carcinoma and non-small cell carcinoma that we might see in lung cancer? It’s not small cell though. These two populations are clearly two adenocarcinomas. That still can’t be. There has to be a population of mesothelials. No, no… the benign mesothelials are scattered throughout the background. It looks like a double-malignant three-cell population!
We submitted the case to the cytopathologist on service for the day. When they asked what we had, we replied, “All the adenocarcinoma,” and noted that we definitely need ER/PR/HER2 as clinically and now morphologically, it appears that the patient’s breast cancer is involving the pleural fluid. The cytopathologist agreed that there was something to the case. Metastatic breast cancer likes to appear as cannon balls in fluid (Image 2), the second cell population is too unlike the first. The cytopathologist performed immunocytochemical stains on paraffin sections of the cell block with adequate controls. The first type of cells shows positive staining for CK7, CK19, GATA3 (Image 5), and ER, and negative staining for CK20 (Image 6), CDX-2 (Image 7), BRST2, and PR. The second type of tumor cells show positive staining for CK7, CK20 (Image 6), CK19, and CDX-2 (Image 7), and negative staining for GATA3 (Image 5), ER, PR, and BRST2. The former group of cells has a morphology and immunoprofile consistent with breast origin, and the latter group of cells are morphologically and immunophenotypically suggestive of a pancreaticobiliary or gastrointestinal tract origin.
Final Diagnosis: Positive for malignancy. Adenocarcinoma, consistent with metastatic breast and metastatic pancreaticobiliary or gastrointestinal tract origin.
Interestingly, the patient had a paracentesis and liver biopsy the following week, and only the pancreaticobiliary or GI tract origin immunoprofile was positive in the peritoneal fluid and FNA. The patient’s breast cancer seemed to remain above the diaphragm.
-Taryn Waraksa-Deutsch, DHSc, SCT(ASCP)CM, CMIAC, LSSGB, is the Cytopathology Supervisor at Fox Chase Cancer Center, in Philadelphia, Pennsylvania. She earned her master’s degree from Thomas Jefferson University in 2014 and completed her Doctorate of Health Science from Bay Path University in 2023. Her research interests include change management and continuous improvement methodologies in laboratory medicine. She is an ASCP board-certified Specialist in Cytology with an additional certification by the International Academy of Cytology (IAC). She is also a 2020 ASCP 40 Under Forty Honoree. Outside of her work, Taryn is a certified Divemaster. Scuba diving in freshwater caverns is her favorite way to rest her eyes from the microscope.