A 78 year old woman was transferred from a nursing home to the Emergency Room because of delirium and worsening bilateral chronic foul-smelling hip wounds. Physical exam was notable for a fever of T103.2°F and purulence from the right hip wound.
Lab results included wbc 17.2K/mm3, hct 26%, platelets 694K/mm3, CRP 9.4 mg/dL, and ESR >130 mm/hr. A pelvic CT scan and X-rays of the hips and femurs showed signs of necrotizing infection, with soft tissue defects over both hips accompanied by subcutaneous fluid, inflammation, gas tracking deep to the femurs, and cortical irregularities of both greater trochanters.
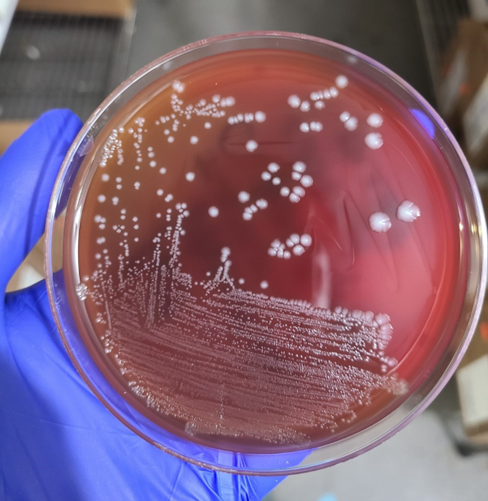
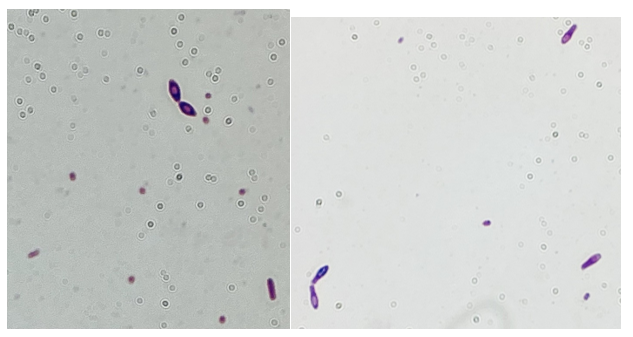
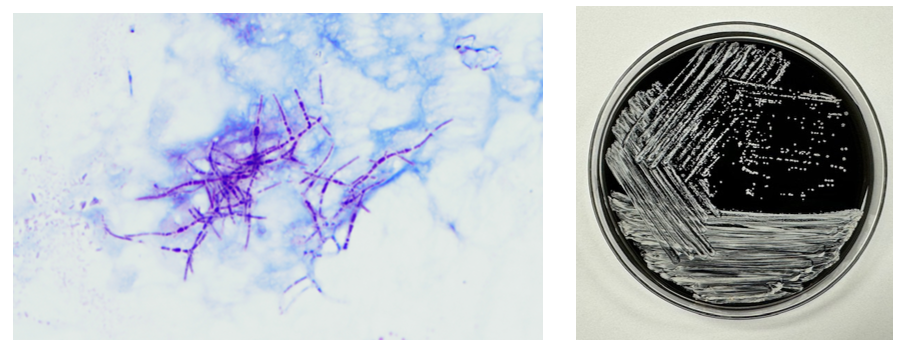
Gram-positive rods grew from the anaerobic bottles of two blood culture sets drawn on arrival at the hospital. Anaerobic blood agar plate growing colonies of gram-positive rods after 48 hours of anaerobic incubation are shown in Figure 1, and Gram stains of the same organism grown in cooked meat broth are shown in Figures 2A and 2B.
Fig 1. Anaerobic CDC blood agar plates growing gram positive bacilliFig 2A and 2B: Gram stain from Cooked meat broth
Gram-positive bacilli (GPB) were isolated from the blood. GPB in blood cultures are often brushed off as possible contaminants. However, in the setting of a possible necrotizing soft tissue infection (NSTI) and the growth of GPB in anaerobic bottles only, concern for Clostridium spp. is reasonable. NSTI can be caused by a variety of different bacteria, but empiric treatment should reliably cover Clostridium species, Streptococcus pyogenes, and Staphylococcus aureus, as they are the most commonly implicated pathogens. While C. perfringens is a common cause for gas gangrene, C. septicum frequently causes non-traumatic gas gangrene because of its aerotolerance.1
The GPB in this patient’s blood was identified by Matrix-assisted laser desorption ionization time of flight mass spectrometry (MALDI-ToF MS) as C. sporogenes/botulinum group I. While C. botulinum is a more familiar pathogen, both of these two closely related bacteria can produce botulinum neurotoxin (BoNT). In a comparative genomic study, C. botulinum Group I was found to possess genes for BoNT A, B, and/or F, while the C. sporogenes possessed BoNT B only.2 Detection of BoNT remains a challenge. Additionally, MALDI-ToF MS cannot distinguish between C. botulinum and C. sporogenes in most cases due to the similarity between these two species.
Since BoNT is considered a category A Biological agent, caution must still be taken when processing suspicious C. botulinum isolates in the laboratory during the identification. While the specimen collection and transport guidelines from American Society of Microbiology (ASM) described not attempting to culture the organism, the guidelines stated that clinical laboratories may still perform routine cultures that may contain Botulinum that potentially produces BoNT.
While it can be challenging to determine the presence of C. botulinum in wound cultures due to the gram feature similarities to skin flora gram positive rods, the laboratories should process the culture workup in biosafety level-2 (BSL-2) cabinet and avoid aerosol-generating procedures (e.g. catalase) to minimize the potential aerosolization of the toxin. The best practice would be an open communication between clinicians and the laboratory – for clinicians to notify the laboratory of potential BoNT cases/cultures when they send microbiology specimens. Post-analytical safety measures must be performed. So, what lesson did we learn here? While it is challenging to distinguish between C. botulinum and C. sporogenes in this case, a proper chain of actions (analytical and post-analytical measurements) should have been taken place to rule out/in BoNT-producing C. botulinum.
Susceptibility testing for anaerobes is not performed routinely and is only appropriately performed on isolates from sterile sources. Globally, rates of clindamycin resistance appear to be increasing among Clostridium spp. However, metronidazole and amoxicillin-clavulanate remain viable options for treatment of Clostridium spp.3-6
Sárvári KP, Rácz NB, Burián K. Epidemiology and antibiotic susceptibility in anaerobic bacteraemia: a 15-year retrospective study in South-Eastern Hungary. Infect Dis (Lond). 2022 Jan;54(1):16-25. doi: 10.1080/23744235.2021.1963469. Epub 2021 Sep 24.
Ali S, Dennehy F, Donoghue O, McNicholas S. Antimicrobial susceptibility patterns of anaerobic bacteria at an Irish University Hospital over a ten-year period (2010-2020). Anaerobe. 2022 Feb;73:102497. Epub 2021 Dec 5.
Di Bella S, Antonello RM, Sanson G, Maraolo AE, Giacobbe DR, Sepulcri C, Ambretti S, Aschbacher R, Bartolini L, Bernardo M, Bielli A, Busetti M, Carcione D, Camarlinghi G, Carretto E, Cassetti T, Chilleri C, De Rosa FG, Dodaro S, Gargiulo R, Greco F, Knezevich A, Intra J, Lupia T, Concialdi E, Bianco G, Luzzaro F, Mauri C, Morroni G, Mosca A, Pagani E, Parisio EM, Ucciferri C, Vismara C, Luzzati R, Principe L. Anaerobic bloodstream infections in Italy (ITANAEROBY): A 5-year retrospective nationwide survey. Anaerobe. 2022 Jun;75:102583. Epub 2022 May 11.
-Antoinette Acobo, PharmD, is 2nd year pharmacy resident specialized in infectious diseases in her 2nd year of residency. She performed several quality initiative and improvement projects, including antimicrobial stewardship program (ASP) and cost/benefit analyses of rapid blood culture identification (BCID) multiplex panels.
-Phyu Thwe, Ph.D, D(ABMM), MLS(ASCP)CM is Associate Director of Infectious Disease Testing Laboratory at Montefiore Medical Center, Bronx, NY. She completed her medical and public health microbiology fellowship in University of Texas Medical Branch (UTMB), Galveston, TX. Her interests includes appropriate test utilization, diagnostic stewardship, development of molecular infectious disease testing, and extrapulmonary tuberculosis.
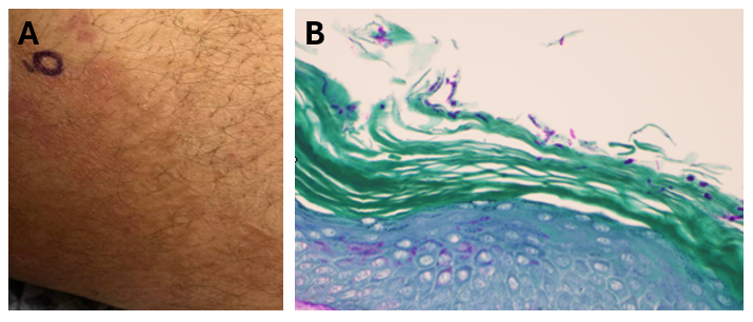
A middle-aged male presented for a chronic inguinal and back rash present for over two years (Figure 1A). Travel history was notable for trips to the Middle East and Canada during that time. He was clinically diagnosed with tinea cruris at an outside healthcare facility, and his history was notable for an extensive use of several topical antifungals including clotrimazole with betamethasone, ketoconazole, and triamcinolone without improvement. Months long oral therapy with terbinafine and fluconazole resulted in only mild improvement, after which he was started on itraconazole. The patient noted some improvement while taking itraconazole, but following subsequent disease recurrence, presented to our institution.
Figure 1: A) Red annular, scaly rash of the inguinal skin involving the inner thigh. Skin biopsy of the thigh rash (40X, PAS stain) revealing dermatophyte hyphal elements in the stratum corneum.
Laboratory workup
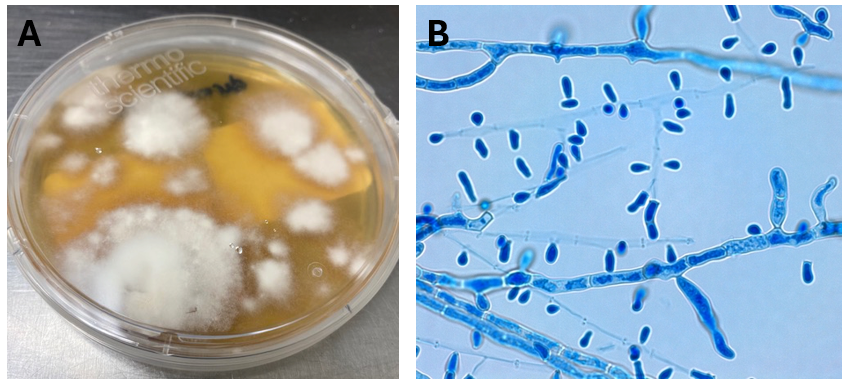
An inguinal skin scraping was obtained, which revealed the presence of a superficial fungal infection consistent with the diagnosis of tina cruris. Fungal cultures on Sabouraud Dextrose Agar (Figure 2A), lactophenol blue stain (Figure 2B), and a skin biopsy (Figure 1B) all confirmed a dermatophyte infection. The organism was morphologically consistent with Trichophyton sp.Due to the unusual clinical history of a recalcitrant dermatophyte infection persisting through multiple rounds of topical and oral antifungal therapy, additional testing for definitive identification was undertaken. MALDI-TOF identified the organism as a member of the Trichophyton tonsurans/mentagrophytes species complex, further confirmed to be Trichophyton indotineae by sequencing.
Figure 2: A) Dermatophyte growth from the skin scraping of the leg on Sabouraud Dextrose agar. Fungal colonies were fast-growing and peripherally white-beige to light brown, flat and granular. B) Lactophenol Cotton Blue stain revealing septate hyphae with cigar-shaped macroconidia, small round microconidia clustered on branched conidiophores.
Discussion
Ringworm, also referred to as “tinea” or “dermatophytosis,” is a commonly occurring fungal infection affecting the skin, hair, or nails, caused by dermatophytes.1 Typical symptoms include itching, a ring-shaped rash, redness, scaliness, cracked skin, and/or hair loss. The transmission of ringworm happens through direct contact between individuals, contact with infected animals, or exposure to contaminated environments such as public showers.1 Around 40 different species of fungi can lead to ringworm infection.2 Over the last decade, healthcare providers have observed a rise in severe cases of ringworm that are resistant to topical antifungal treatment3 which are an emerging public health concern.
The emergence of drug-resistant dermatophyte infections is attributed to the inappropriate use of topical products containing combinations of antifungal agents and corticosteroids.4T. indotiniae has a complicated taxonomic history. The organism was initially identified as a sequence type of the Trichophyton mentagrophytes complex which exhibited elevated MICs to terbinafine. The earliest cases of T. indotineae infection were reported in India, quickly spreading to Australia, Oman, Iran, and other middle-eastern countries. Subsequent spread to Europe and North America soon followed.5 Initial reports of T. indotineae infections in North America involved individuals with a history of travel to endemic locations. Recently however, local transmission of T. indotineae within the United States has been documented among patients without travel history.6 It is hypothesized that resistant strains of T. indotineae emerged due to inappropriate antibiotic use, as infections are frequently terbinafine-resistant and require prolonged therapies with second-line therapies or antifungals traditionally utilized for invasive fungal infections.7
Diagnosis of T. indotineae infection is challenging as it requires advanced molecular techniques like genomic sequencing or expansion of MALDI-TOF MS capabilities to discriminate within the T. mentagrophytes complex, which many clinical laboratories lack.1 Thus, diagnosis is largely reliant on the activities of reference laboratories. A high level of clinical suspicion is required as well, as the degree of dermatophyte workup undertaken in the routine setting is variable among institutions. T. indotineae infections often show resistance to conventional antifungal therapies including allylamines (terbinafine) and azoles (itraconazole and fluconazole), further highlighting the importance of susceptibility testing before initiating treatment and reassessing nonresponsive cases,4 another testing capability not offered by routine laboratories. The patient in this case was managed with Posaconazole, leading to significant improvements symptomology.
Gupta AK, Venkataraman M, Hall DC, Cooper EA, Summerbell RC. The emergence of Trichophyton indotineae: Implications for clinical practice. Int J Dermatol. 2023 Jul;62(7):857-861. doi: 10.1111/ijd.16362. Epub 2022 Jul 22. PMID: 35867962.
Jia S, Long X, Hu W, Zhu J, Jiang Y, Ahmed S, de Hoog GS, Liu W, Jiang Y. The epidemic of the multiresistant dermatophyte Trichophyton indotineae has reached China. Front Immunol. 2023 Feb 16;13:1113065. doi: 10.3389/fimmu.2022.1113065. PMID: 36874152; PMCID: PMC9978415.
Caplan AS, Chaturvedi S, Zhu Y, Todd GC, Yin L, Lopez A, Travis L, Smith DJ, Chiller T, Lockhart SR, Alroy KA, Greendyke WG, Gold JAW. Notes from the Field: First Reported U.S. Cases of Tinea Caused by Trichophyton indotineae – New York City, December 2021-March 2023. MMWR Morb Mortal Wkly Rep. 2023 May 12;72(19):536-537. doi: 10.15585/mmwr.mm7219a4. PMID: 37167192; PMCID: PMC10208369.
Lockhart, SR, Smith, DJ, and Gold, J.A.W. Trichophyton indotineae and other terbinafine-resistant dermatophytes in North America. J. Clin. Microbiol. 2023 Dec; 61(12): e00903-23. PMID: 38014979.
–Tasnim Alkayyali is a second-year AP/CP resident at UT Southwestern Medical Center in Dallas, Texas.
–Clare McCormick-Baw, MD, PhD, FACP is a board certified Anatomic and Clinical Pathologist with a subspecialty in Medical Microbiology. She has a love for Infectious Disease Pathology and teaching the pathologists of tomorrow. She is the Southwest Regional Medical Director for Quest Diagnostics, Inc and is based out of Dallas, Texas.
–Andrew Clark, PhD, D(ABMM) is an Assistant Professor at UT Southwestern in the Department of Pathology and Director of the Microbiology Laboratory at Parkland Health and Hospital System. He completed a CPEP-accredited postdoctoral fellowship in Medical and Public Health Microbiology at National Institutes of Health.
A 73 year old male with a complex medical history, including type 1 diabetes, coronary artery disease, atrial fibrillation, hypothyroidism, hyperlipidemia, glaucoma, previous coronary artery bypass grafting (CABG), transcatheter aortic valve replacement (TAVR), and an implanted cardioverter-defibrillator (ICD), presented with bilateral persistent paraspinal neck pain, shaking chills, and myalgias. The initial evaluation involved broad laboratory and imaging work-up, which showed mildly elevated CRP and erythrocyte sedimentation rate (ESR). Chest X-ray, CTA head/neck, and CT thorax were normal. The patient was discharged with symptoms resolved using Tylenol and Valium, and instructions for follow-up care were provided. Two days later, the patient was recalled to the ED after blood cultures were positive for gram positive cocci in pairs and chains. The patient, asymptomatic at the time, was informed of the findings and agreed to return for further evaluation.
The patient had undergone a tooth extraction followed by implant placement one month prior, raising concerns for endocarditis due to prosthetic valve and pacemaker. He was transferred to the cardiology service, and repeat cultures were ordered. Initially afebrile, he later developed a fever (101.7°F), and his condition was complicated by diabetic ketoacidosis (DKA) and septic shock requiring ICU admission. The patient was treated with IV Vancomycin 1.5 g q24h and recommended to have repeat blood cultures until clearance. The discharge diagnosis was bacteremia caused by Abiotrophia defectiva, with a possible prosthetic valve endocarditis. A six-week course of Vancomycin was prescribed.
(A) Gram stain of the positive blood culture bottle revealed gram positive cocci in chains and pairs. (B) Inoculation of the positive blood culture bottle onto bacterial agar plates showed no growth on sheep’s blood agar but growth on chocolate agar.
Discussion
The genus Abiotrophia most resembles well-known gram positive cocci in chains and pairs such as streptococci and enterococci. Abiotrophia were originally thought to be a relative of the viridans Group Streptococcus but sequencing analysis created a new genus called Abiotrophia which consisted of two species, A. defective and A. adiacens.1Abiotrophia is classified as nutritionally variant streptococcus (NVS) and is a part of normal flora in the intestinal tract, oral cavity, and urogenital tract. The organism accounts for up to 6% of streptococcal infective endocarditis and have been associated with native and prosthetic valve infection.2 Other types of infections that have been described for Abiotrophia include sepsis, infections of the eye, central nervous system, musculoskeletal, and arthritis.3-6 It is crucial to recognize A. defectiva for timely and effective treatment, given its propensity to cause severe complications like septic embolization.7
Abiotrophia is coined a NVS because this microorganism requires specific nutrients like L-cysteine or pyridoxal for growth on sheep’s blood agar. They can usually grow without supplementation on chocolate agar, brucella agar with 5% horse blood, and in thioglycolate broth. Sometimes sheep’s blood agar supplemented with pyridoxal can also be used. Due to the release of specific nutrients by lysed red blood cells, the satellite test is important for growth and identification. Briefly, the suspected isolate is streaked for confluent growth on an agar that does not support growth for Abiotrophia (such as sheep’s blood agar) and then overlaid with a cross streak of beta-hemolytic Staphylococcus aureus. After incubation, colonies of Abiotrophia can grow near the S. aureus streak, giving rise to the ‘satellite’ phenomenon. However, it should be noted that other fastidious organisms such as Granulicatella and some Haemophilus species can also satellite around the S. aureus streak. Hence, further confirmation is needed for identification. For example, Abiotrophia is non-motile. Common biochemicals reactions to help characterize Abiotrophia genus is PYR (production of pyrrolidonyl arylamidase) positive, LAP (production of leucine aminopeptidase) positive, and no growth in 6.5% NaCl.
Molecular approaches have been proven to be accurate for identification of Abiotrophia. Sequencing of the 16S rRNA gene have shown to be more accurate than phenotypic, biochemical tests. PCR testing for the rpoB, groESL, and also the ribosomal 16S-23S intergenic spacer region (ITS) genes have all shown to be reliable targets for identification.8-10 The introduction of MALDI-TOF mass spectrometry has facilitated more accurate identification in clinical settings.11
Prompt diagnosis and targeted antimicrobial therapy are essential in managing infections caused by Abiotrophia defectiva, especially in older patients with underlying health conditions. Early detection and appropriate treatment are vital to mitigate potential complications.6,7Abiotrophia species exhibit varying antibiotic susceptibility, with increasing resistance to penicillin. Common treatments include penicillin, ceftriaxone, and vancomycin.
References
1. Kawamura, Y., et al., Transfer of Streptococcus adjacens and Streptococcus defectivus to Abiotrophia gen. nov. as Abiotrophia adiacens comb. nov. and Abiotrophia defectiva comb. nov., respectively. Int J Syst Bacteriol, 1995. 45(4): p. 798-803.
2. Christensen, J.J. and R.R. Facklam, Granulicatella and Abiotrophia species from human clinical specimens. J Clin Microbiol, 2001. 39(10): p. 3520-3.
3. Niu, K. and Y. Lin, Abiotrophia defectiva Endocarditis Complicated by Stroke and Spinal Osteomyelitis. Cureus, 2024. 16(3): p. e56904.
4. Wilhelm, N., et al., First case of multiple discitis and sacroiliitis due to Abiotrophia defectiva. Eur J Clin Microbiol Infect Dis, 2005. 24(1): p. 76-8.
5. Taylor, C.E. and M.A. Fang, Septic arthritis caused by Abiotrophia defectiva. Arthritis Rheum, 2006. 55(6): p. 976-7.
6. Yang, M., et al., Abiotrophia defectiva causing infective endocarditis with brain infarction and subarachnoid hemorrhage: a case report. Front Med (Lausanne), 2023. 10: p. 1117474.
7. Faria, C., et al., Mitral Valve Subacute Endocarditis Caused by Abiotrophia Defectiva: A Case Report. Clin Pract, 2021. 11(1): p. 162-166.
8. Drancourt, M., et al., rpoB gene sequence-based identification of aerobic Gram-positive cocci of the genera Streptococcus, Enterococcus, Gemella, Abiotrophia, and Granulicatella. J Clin Microbiol, 2004. 42(2): p. 497-504.
9. Hung, W.C., et al., Use of groESL as a target for identification of Abiotrophia, Granulicatella, and Gemella species. J Clin Microbiol, 2010. 48(10): p. 3532-8.
10. Tung, S.K., et al., Identification of species of Abiotrophia, Enterococcus, Granulicatella and Streptococcus by sequence analysis of the ribosomal 16S-23S intergenic spacer region. J Med Microbiol, 2007. 56(Pt 4): p. 504-513.
11. Christensen, J.J., et al., Matrix-assisted laser desorption ionization-time of flight mass spectrometry analysis of Gram-positive, catalase-negative cocci not belonging to the Streptococcus or Enterococcus genus and benefits of database extension. J Clin Microbiol, 2012. 50(5): p. 1787-91.
-Maher Ali, MD was born in and raised in Amman, Jordan. He attended Jordan University of Science and Technology, where he received his doctorate degree. After graduation, he completed two years of anatomic pathology residency at King Hussein Cancer Center in Jordan. He then relocated to the United States to start his combined anatomic and clinical pathology (AP/CP) training at The George Washington University. His academic interests include gastrointestinal and breast pathology. Outside of the hospital, he enjoys cooking, hiking, exploring new restaurants, and spending quality time with his family and friends.
-Rebecca Yee, PhD, D(ABMM), M(ASCP)CM is the Chief of Microbiology, Director of Clinical Microbiology and Molecular Microbiology Laboratory at the George Washington University Hospital. Her interests include bacteriology, antimicrobial resistance, and development of infectious disease diagnostics. She is also a top 5 honoree among the 2023 ASCP 40 Under Forty.
A 36 year old male with no past medical history presented to an outside hospital with two weeks of worsening headache, fever, and altered mental status. The patient initially presented to his primary care physician with a headache, for which he received an antibiotic injection. Over the next several days, he developed a fever to 39.4oC, worsening mental status, and bowel incontinence. Once admitted, he experienced an intractable seizure, was subsequently intubated, and transferred to the ICU. CT imaging revealed multiple ring-enhancing lesions throughout the brain, most prominent in the right parietal lobe. Additionally, he was found to have a new diagnosis of HIV (CD4 count 40). He was transferred to our institution for a higher level of care but experienced a rapid progression of disease with deterioration of brainstem reflexes and marked cerebral edema, prompting a brain biopsy.
Laboratory Identification
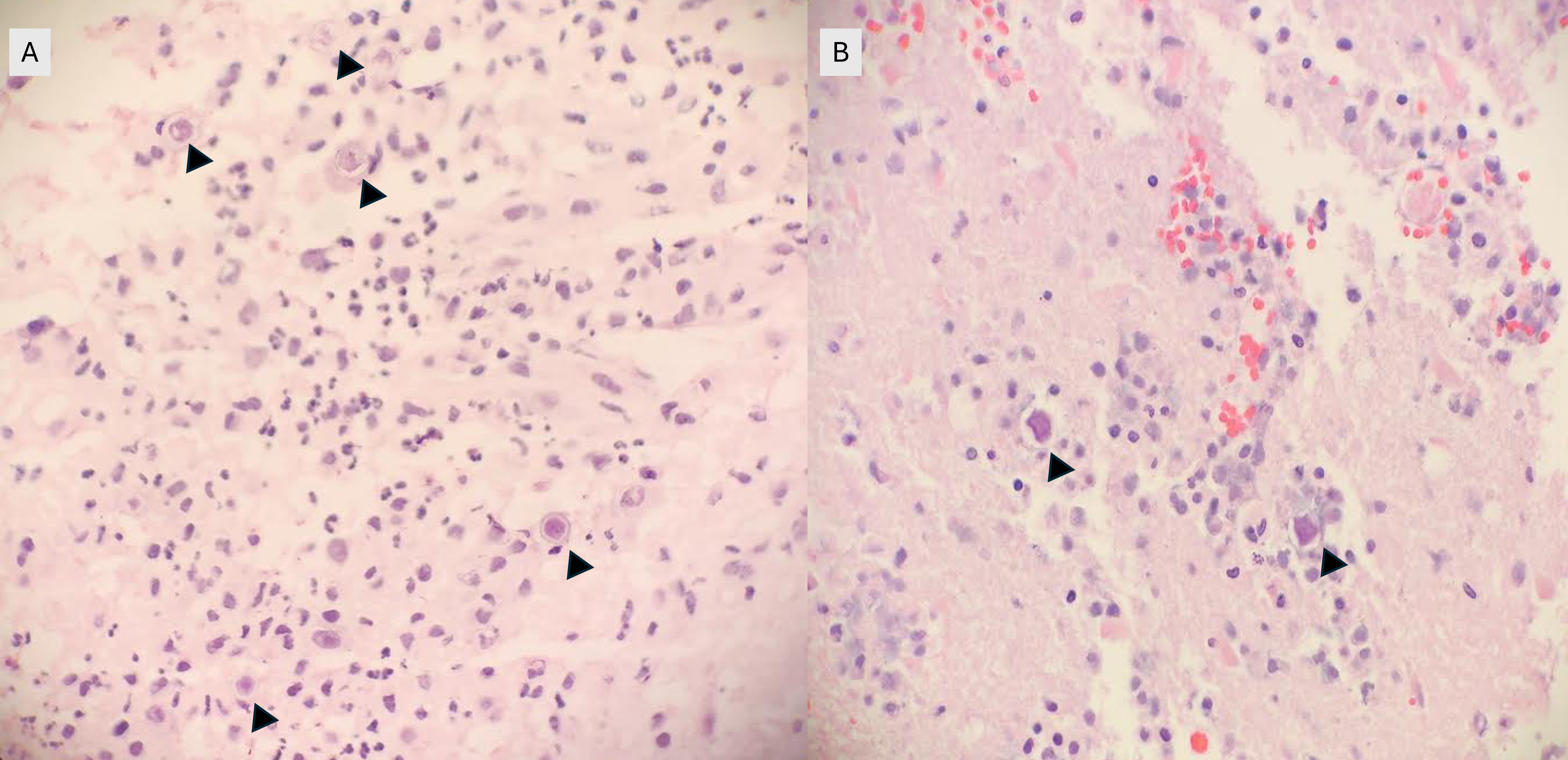
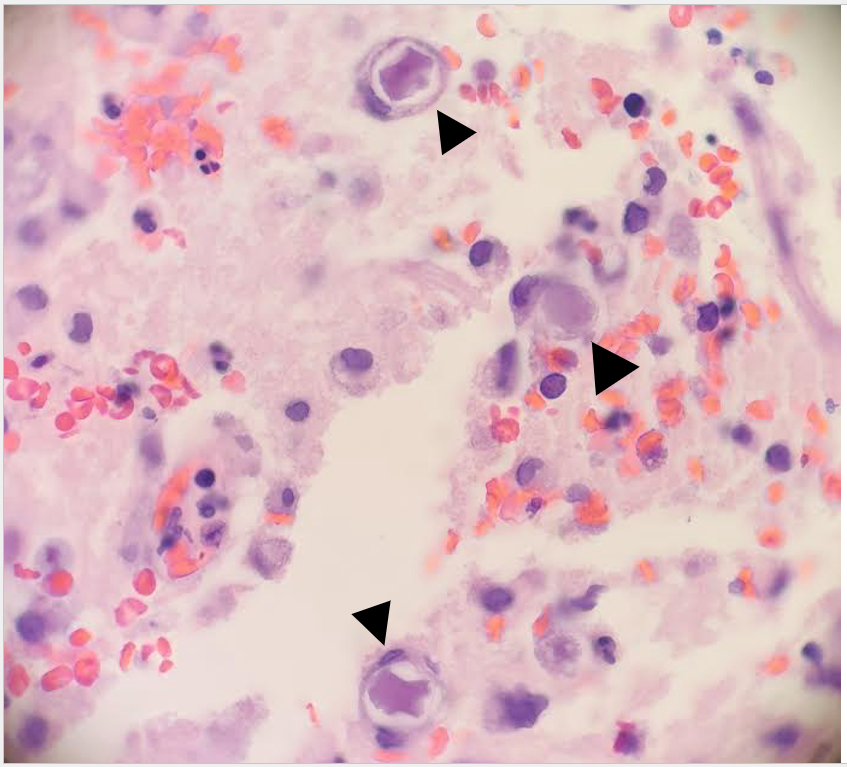
Frozen and permanent sections of a right temporal biopsy revealed frankly necrotic brain tissue with amoebic cysts and trophozoites, consistent with free-living amoeba (Figure 1 and 2). Cerebrospinal fluid (CSF) cytology was unremarkable. A specific immunohistochemistry (IHC) assay for Acanthamoeba species was performed at the Centers for Disease Control and Prevention (CDC; Atlanta, GA), confirming the diagnosis of granulomatous amoebic encephalitis from Acanthamoeba.
Figure 1. Frozen (A) and permanent (B) sections of a right temporal brain biopsy demonstrated amoebic cysts (arrowheads) in a background of necrosis, hemorrhage, and inflammation (H&E, 400x magnification).Figure 2: Double-walled cysts (arrowheads) are observed in the brain biopsy with admixed inflammation and hemorrhage (H&E, 1000x oil immersion magnification).
Discussion
Acanthamoeba are free-living amoebae found worldwide in a variety of environmental sites, including soil, water, sewage, swimming pools, contaminated contact lens solution, and heating, ventilating, and air conditioning systems.1,2 The Acanthamoeba lifecycle consists of two stages: cysts and trophozoites. Although trophozoites represent the infective form, both cysts and trophozoites can enter the body through various means, including the eye, nasal passages, or broken skin.2,3. There are three distinct diseases that can be caused by Acanthamoeba: Acanthamoeba keratitis, disseminated infection, and Granulomatous Amebic Encephalitis (GAE).1
GAE is a parenchymal disease that occurs when Acanthamoeba enters the body through the respiratory system or broken skin, spreads hematogenously, and invades the central nervous system.2,3. Risk factors include immunocompromised status, such as HIV/AIDS, history of organ transplant, uncontrolled diabetes mellitus, liver cirrhosis, and malignancy.1,3 The clinical course tends to be subacute to chronic, with progressive neurological decompensation over weeks to months and eventual death. Imaging may reveal ring-enhancing lesions, arterial occlusions, infarctions, ventricular shifts, and areas of abnormal enhancement.3 CSF analyses may show a pleocytosis with lymphocyte predominance, increased protein, and decreased glucose; amoebae are typically not identified.3. While microscopic evidence of double-walled cysts and/or trophozoites demonstrates the presence of free-living amoeba in infected tissues, diagnosis of Acanthamoeba must be confirmed via specific IHC assays or PCR-based molecular techniques.4
There is currently no standardized recommended treatment, and no single agent has been shown to be universally effective.3. Rare successful cases have involved prolonged therapy using combinations of pentamidine isethionate, sulfadiazine, amphotericin D, azithromycin, flucytosine, and fluconazole or itraconazole.3,5
The patient was treated with miltefosine, fluconazole, pentamidine, flucytosine, and sulfadiazine, sulfamethoxazole/trimethoprim and azithromycin for HIV prophylaxis, and levetiracetam for seizure prophylaxis. Though repeat MRI of the brain/brain stem showed interval improvement of disease burden, he remained unresponsive to verbal, tactile, and painful stimuli, and his overall prognosis remained guarded. The patient was discharged to a long-term acute care facility, but returned to the emergency department one week later with recurrent seizures, hypotension, and fevers. Imaging revealed new areas of restricted diffusion in the right occipital and left parietal lobes. Despite continued treatment, the patient continued to decline and died approximately three months following his initial diagnosis.
3 Siddiqui R, Khan NA. Biology and pathogenesis of Acanthamoeba. Parasit Vectors. 2012 Jan 10;5:6. doi: 10.1186/1756-3305-5-6. PMID: 22229971; PMCID: PMC3284432.
4 Haldar SN, Banerjee K, Modak D, Mondal A, Sharma C, Vasireddy T, Karad RK, Patel HB, Majumdar D, Bhattacharjee B, Khurana S, Ghosh T, Guha SK, Saha B. Case Report: A Series of Three Meningoencephalitis Cases Caused by Acanthamoeba spp. from Eastern India. Am J Trop Med Hyg. 2024 Jan 9;110(2):246-249. doi: 10.4269/ajtmh.23-0396. PMID: 38190743; PMCID: PMC10859797.
-Cristine Kahlow, MD, Pathology PGY-1 at UTSW Medical Center
-Clare McCormick-Baw, MD, PhD is an Assistant Professor of Clinical Microbiology at UT Southwestern in Dallas, Texas. She has a passion for teaching about laboratory medicine in general and the best uses of the microbiology lab in particular.
-Andrew Clark, PhD, D(ABMM) is an Assistant Professor at UT Southwestern Medical Center in the Department of Pathology, and Associate Director of the Clements University Hospital microbiology laboratory. He completed a CPEP-accredited postdoctoral fellowship in Medical and Public Health Microbiology at National Institutes of Health, and is interested in antimicrobial susceptibility and anaerobe pathophysiology
A 70 year old female with a medical history of treatment-related acute myeloid leukemia with absolute neutropenia due to chemotherapy presented to our emergency department with a two-day history of fever (Tmax 102°C) with progressively increasing erythema and tenderness reported in her right elbow around a healing surgical incision. Approximately one month prior to presentation the patient underwent surgical excision of her right upper extremity (RUE) cephalic vein for supportive thrombophlebitis as a complication of peripheral IV insertion. Additionally, two weeks prior to presentation the patient was noted to have three small areas (< 1 cm) of wound dehiscence along the surgical incision without overt signs of infection at her outpatient surgical follow-up visit (Figure 1A). These wounds were packed with iodoform gauze and wrapped with sterile kerlix gauze wrap followed by an elastic wrap. The patient was instructed to change the packing and dressing one to two times daily; however, the patient reported non-compliance with these instructions and the packing and dressing had not been changes in the two weeks from the clinic visit until ED presentation.
Figure 1. Images of right upper extremity wound. Image A was obtained two weeks prior to presentation at an outpatient surgical follow-up appointment showing three small areas of wound dehiscence (black arrows) but no overt signs of infection. Image B was obtained at presentation and reveals the three areas of wound dehiscence filled with old iodoform gauze with erythema surrounding the surgical incision. Image C taken ~24 hours after image B shows worsening and progression of the cutaneous erythema surrounding the wound.
The patient was admitted to the hospital. Blood cultures were drawn and IV vancomycin, cefepime and metronidazole were initiated for empirical antimicrobial therapy in the setting of neutropenic fever and RUE cellulitis. 18 hours after collection both sets of blood cultures grew a gram positive rod. The infection diseases service was subsequently consulted to determine the significance of the gram positive rods growing in blood culture, if it represented contaminant vs a true pathogen. Imaging of the arm was recommended to further evaluate the extent of the RUE skin & soft tissue infection. CT imaging of the humerus and forearm revealed a thick-walled fluid collection concerning for abscess formation along the operative site involving both the arm and forearm of the patient (Fig. 2). The plastic surgery service was also consulted and noted old iodoform gauze in the areas of wound dehiscence with surrounding erythema (Fig. 1B); however, no purulence was noted from these areas. The erythema surrounding the wound continued to progress (Fig. 1C) during the hospitalization and the patient remained febrile despite the broad-spectrum antibiotic regiment. Given the patient treatment failure by medical management, the patient was taken for surgical washout and debridement of the wound. Deep cultures were obtained during the surgery. Both the tissue culture and blood cultures would grow Corynebacterium jeikeium. The patient developed a maculopapular cutaneous rash several days into the admission which was deemed to be an antibiotic-induced rash. The antimicrobial regiment was narrowed down to daptomycin due to the concern that vancomycin and/or cefepime may be responsible for the drug rash. The cellulitis and abscess resolved following surgical debridement and treatment with vancomycin/daptomycin; however, the patient elected to pursue comfort care status due to her underlying malignancy. She was transitioned to inpatient hospice where she would die shortly afterwards due to progression of her AML.
Figure 2.Images of the computed topography (CT) with contrast from RUE. Views of the humerus (Image A) and forearm (Image B) revealed likely abscess formation (white arrows) measuring approximately 6.8 cm in length and approximately 1.5 cm in greatest dimension in both the arm (humerus) and forearm.
Laboratory
Corynebacterium species are non-motile, facultatively anaerobic, club-shaped gram positive rods that have a characteristic picket fence appearance when stained due to snapping division. These organisms are commonly encountered in clinical samples; however, given their low virulence in general and prevalence as common skin flora they are often considered contaminants or colonizers. The clinical significance of diphtheria toxin (DT)-producing strains has been known for decades. Non-DT-producing Corynebacterium species were historically considered as non-virulent but in recent years several non-DT-producing Corynebacterium species have been noted to be pathogenic. The wide spread availability of MALDI-TOF MS in most clinical microbiology labs has allowed for rapid and accurate species level identification of Corynebacterium allowing for identification of these pathogenic species in clinical settings. Some Corynebacterium species are highly lipophilic due to the presence of fatty acids, such as mycolic acid, in their cell wall. Isolation of lipophilic Corynebacterium species on standard culture media can be challenging but can be accomplished utilizing blood agar, due to the presence of lipid-containing red cell membranes, with at least 48 hours of incubation. For optimal recovery of lipophilic strains agars supplemented with Tween 80 produce the best recovery rates.
As a lipophilic species, C. jeikeium can be difficult to identify and propagate. It does not grow well on chocolate agar (Fig. 3A & 3C). On blood agar it tends to have scant or dusky growth at 24 hours (Fig. 3B) and appear as translucent, pinpoint colonies at 48 hours (Fig. 3D). Identification using MALDI-TOF can be challenging before 48 hours due to its poor growth on standard media. For comparison, C. striatum, a non-lipophilic Corynebacterium spp. grows relatively well on both blood and chocolate agar. Several Corynebacterium species, including C. jeikeium and C. striatum, are known to be multidrug resistant, especially towards beta-lactam antibiotics.
Figure 3.Images of secondary culture of C. jeikeium and C. striatum inoculated onto chocolate agar (left column) and blood agar (right column) and incubated aerobically at 35°C for 24-48 hours.
Discussion
Corynebacterium jeikeium was initially identified in 1976 and classified by CDC as group JK diptheroids. It is considered part of normal skin flora like most other Corynebacterium spp. In immunosuppressed individuals, especially in persons with hematolymphoid malignancies, invasive disease including dissemination infections have been reported. Infections can also be seen in hardware-associated and prosthetic joint-related infections. Due to its predilection for biofilm formation, C. jeikeium can be difficult to treat in these cases. Cases of infective endocarditis have been reported as well. Like many Corynebacterium species, C. jeikeium is often multidrug resistant. Vancomycin is the first line agent for treatment of infections due to C. jeikeium. Linezolid and daptomycin are other alternative treatment options. When isolating C. jeikeium from blood sources especially in immunocompromised patients like in our case, physicians and other health-care providers must thoroughly assess the total clinical picture before dismissing it as a contaminant.
References
1. Bernard K., Identification of Gram-Positive Bacteria (2023) in AL Leber & CAB Burnham (Eds.) Clinical Microbiology Procedures Handbook (5th ed.) doi:10.1128/9781683670438.CMPH.ch3.18
2. Gupta R, Popli T, Ranchal P, Khosla J, Aronow WS, Frishman WH, El Khoury MY. Corynebacterium Jeikeium Endocarditis: A Review of the Literature. Cardiol Rev. 2021 Sep-Oct 01;29(5):259-262. doi: 10.1097/CRD.0000000000000355. PMID: 32976125.
3. Mattos-Guaraldi AL, Sanchez Dos Santos L, & Vieira VV, Coryneform Gram-Positive Rods (2023) in Carroll et al. (Eds.) Manual of Clinical Microbiology (15th ed.) doi:10.1128/9781683670438.MCM.ch28
4. Moore Pardo SM, Patel RH, Ramsakal A, Greene J. Disseminated Corynebacterium jeikeium Infection in Cancer Patients. Cureus. 2020 Jun 22;12(6):e8764. doi: 10.7759/cureus.8764. PMID: 32714702; PMCID: PMC7377673.
5. Murray BE, Karchmer AW, Moellering RC Jr. Diphtheroid prosthetic valve endocarditis. A study of clinical features and infecting organisms. Am J Med. 1980 Dec;69(6):838-48. doi: 10.1016/s0002-9343(80)80009-x. PMID: 7446550.
6. Ozdemir S, Aydogan O, Koksal Cakirlar F. Biofilm Formation and Antimicrobial Susceptibility of Non-Diphtheria Corynebacterium Strains Isolated from Blood Cultures: First Report from Turkey. Medeni Med J. 2021;36(2):123-129. doi: 10.5222/MMJ.2021.60252. Epub 2021 Jun 18. PMID: 34239764; PMCID: PMC8226407.
7. Tauch A, Kaiser O, Hain T, Goesmann A, Weisshaar B, Albersmeier A, Bekel T, Bischoff N, Brune I, Chakraborty T, Kalinowski J, Meyer F, Rupp O, Schneiker S, Viehoever P, Pühler A. Complete genome sequence and analysis of the multiresistant nosocomial pathogen Corynebacterium jeikeium K411, a lipid-requiring bacterium of the human skin flora. J Bacteriol. 2005 Jul;187(13):4671-82. doi: 10.1128/JB.187.13.4671-4682.2005. PMID: 15968079; PMCID: PMC1151758.
-Arooj Devi MD is a second-year pathology (AP/CP) resident at Brody School of Medicine at East Carolina University. She is interested in breast and gynecological pathology.
-John E. Markantonis DO D(ABMM) is the head of microbiology at ECU Health Medical Center and an assistant professor at Brody School of Medicine at East Carolina University.
A 28 year old woman underwent an elective myomectomy for menorrhagia caused by fibroids. Postoperatively, she developed fevers. A CT scan of the abdomen and pelvis showed a complex pelvic fluid collection measuring 5.4 by 4.4 by 7.0 cm. Drainage was attempted by Interventional Radiology without success. She then underwent an exploratory laparotomy with drainage of the collection, evacuation of a hematoma, and removal of an IUD. The abscess fluid was sent to the Microbiology lab for culture.
The Gram stain of the fluid revealed 1+PMN with no organisms. After 48 hours of incubation, there was few to moderate growth of pinpoint clear colonies on blood agar, with characteristic miniscule appearance of a central area of dense growth and peripherally less dense (Figure 1). Upon closeup reviewing of the colonies revealed a “fried egg” appearance (Fig 2).
There was no growth on MacConkey plate. Final identification by MALDI-TOF demonstrated Mycoplasma hominis.
Figure 1. Blood agar plate growing clear small colonies of M. hominis.Figure 2. Characteristic colonies of organism with peripheral growth and denser central growth with fried-egg appearance.
Discussion
Mycoplasma hominis is a facultative anaerobe of the Mycoplasma genus, which is among the smallest free-living organisms known. They are fastidious and differentiated from other bacteria by their small size and absence of cell walls. Infections with M. hominis predominately involve the urogenital tract of females, causing pelvic inflammatory disease, bacterial vaginosis, and postpartum fever.1 Extragenital manifestations of M. hominis infections are rare but include extragenital abscesses, CNS infections in neonates, and septic arthritis.2 Reports of Mycoplasma hominis infections vary based on demographics (country, age, and number of sexual partners) but have been isolated in 4 to 30% of urogenital infections in females. 1,3
The lack of cell walls confers both diagnostic and clinical challenges. Mycoplasmas is not seen on the routine Gram stain smears. Instead of a cell wall, they have a triple-layered membrane composed of sterol.3 When able to grow in culture, colonies are small and have a ‘fried egg’ appearance on agar.4 Of the Mycoplasma species, M. hominis is the least fastidious and the most common Mycoplasma isolated on conventional culture agar media. 4-6 Non-traditional culture-based diagnosis is often made via molecular testing. 7 It is often detected in coinfections with Trichomonas vaginalis and thought to be a symbiotic relationship. 8-9
Since M. hominis is also found to be colonized in the genital tract of normal healthy individuals, it can be challenging to establish the clinical and diagnostic significance of M. hominis. With evolving molecular diagnostic assays targeting STI agents, it has been a controversial topic for assay manufacturers or labs that develop their own in-house assays whether there is a clinical utility for M. hominis PCR as part of STI multiplex PCR panels.
In contrast to Mycoplasma pneumoniae, M. hominis has intrinsic resistance to macrolides. 10 Preferred treatment regimens for M hominis infections include tetracyclines, clindamycin and fluoroquinolones. 11 However, resistance to members of these antibiotic classes has been reported and differs based on country of origin. 11 The absence of a cell wall explains the inherent resistance of Mycoplasma species to the beta-lactam antibiotic class. Its isolation in coinfections, particularly Trichomonas has been controversial in its contribution to emerging Trichomonas metronidazole resistance. 12
References
Verteramo, R., Pastella, A., Calzolari, E., et al. An epidemiological survey of Mycoplasma hominis and Ureaplasma urealyiticum in gynaecological outpatients, Rome, Italy. Epidemiology & Infection,2013, 141(12), 2650-2657
Zheng X, Olson DA, Tully JG, Watson HL, Cassell Gh, Gustafson DR, Svien KA, Smith TF: Isolation of Mycoplasma hominis from a brain abscess. J Clin Microbiol. 1997, 35: 992-994.
Thomas Prescott Atkinson, Mitchell F. Balish, Ken B. Waites, Epidemiology, clinical manifestations, pathogenesis and laboratory detection of Mycoplasma pneumoniae infections, FEMS Microbiology Reviews, Volume 32, Issue 6, November 2008, Pages 956–973
Razin S. Mycoplasmas. In: Baron S, editor. Medical Microbiology. 4th edition. Galveston (TX): University of Texas Medical Branch at Galveston; 1996. Chapter 37.
Meloni GA, Bertoloni G, Busolo F, Conventi L. Colony morphology, ultrastructure and morphogenesis in Mycoplasma hominis, Acholeplasma laidlawii and Ureaplasma urealyticum. J Gen Microbiol. 1980;116(2):435-443.
Christiansen G, Jensen LT, Boesen T, Emmersen J, Ladefoged SA, Schiotz LK, Birkelund S: Molecular biology of Mycoplasma. Wien Klin Wochenschr. 1997, 109: 557-561
Baczynska, A., Svenstrup, H.F., Fedder, J. et al. Development of real-time PCR for detection of Mycoplasma hominis. BMC Microbiol 4, 35 (2004).
Tine RC, Dia L, Sylla K, Sow D, Lelo S, Ndour CT. Trichomonas vaginalis and Mycoplasma infections among women with vaginal discharge at Fann teaching hospital in Senegal. Trop Parasitol. 2019 Jan-Jun;9(1):45-53.
Vancini, R.G., Benchimol, M. Entry and intracellular location of Mycoplasma hominis in Trichomonas vaginalis . Arch Microbiol 189, 7–18 (2008).
Pereyre S, Gonzalez P, De Barbeyrac B, Darnige A, Renaudin H, Charron A, Raherison S, Bébéar C, Bébéar CM. Mutations in 23S rRNA account for intrinsic resistance to macrolides in Mycoplasma hominis and Mycoplasma fermentans and for acquired resistance to macrolides in M. hominis. Antimicrob Agents Chemother. 2002 Oct;46(10):3142-50.
Krausse R, Schubert S. In-vitro activities of tetracyclines, macrolides, fluoroquinolones and clindamycin against Mycoplasma hominis and Ureaplasma ssp. isolated in Germany over 20 years. Clin Microbiol Infect. 2010;16(11):1649-1655.
Dessì, D., Margarita, V., Cocco, A., Marongiu, A., Fiori, P., & Rappelli, P. (2019). Trichomonas vaginalis and Mycoplasma hominis: New tales of two old friends. Parasitology,146(9), 1150-1155. doi:10.1017/S0031182018002135
-Dr. Alex Shaffer is a first-year ID fellow (2023-2025) at Montefiore Einstein. Alex is interested in the diagnostic stewardship projects and has been actively involved in various activities with ID and micro lab faculty.
-Phyu Thwe, Ph.D, D(ABMM), MLS(ASCP)CM is Associate Director of Infectious Disease Testing Laboratory at Montefiore Medical Center, Bronx, NY. She completed her medical and public health microbiology fellowship in University of Texas Medical Branch (UTMB), Galveston, TX. Her interests includes appropriate test utilization, diagnostic stewardship, development of molecular infectious disease testing, and extrapulmonary tuberculosis.
A 53 year old male presented to the emergency department with a one-day history of malaise, bilateral flank pain and decreased urine output. His past medical history was notable for decompensated cirrhosis due to alcohol use disorder complicated by esophageal varices and gastric ulcers, peritoneal ascites, several recent episodes of upper GI bleeding, monoclonal gammopathy of unknown significance, and remote prostate cancer status post prostatectomy. Oral history indicated the patient was actively drinking shortly before presentation and had recently consumed oysters on the half shell, broiled shrimp, and crab meat. While being seen in the emergency department, the patient quickly progressed to septic shock.
Admission laboratory studies demonstrated severe metabolic acidosis, pancytopenia, and acute renal failure. Initial CT abdomen/pelvis demonstrated cirrhotic liver changes, varices, distal esophageal wall thickening, bilateral perinephric fat stranding extending into the pelvis and perivesical fat. The patient was admitted to the MICU for intubation/mechanical ventilation and administration of multiple pressors, as well as empiric meropenem, vancomycin and micafungin given concern for septic shock.Initial blood cultures drawn in the ED flagged positive with gram negative rods (both aerobic and anaerobic bottles) less than 24 hours after collection.
Laboratory Identification
Blood cultures were processed for culture workup and molecular identification. No identification was able to be established using a commercial multiplex PCR-based molecular panel on the positive blood culture broth. Subsequent growth of the organism on MacConkey agar 18 hours later revealed non-lactose fermenting colonies (Image 1A). The organism was oxidase-negative, positive for catalase and indole (Image 1B) and exhibited hydrogen sulfide production when inoculated on triple sugar iron (TSI) media. The organism was definitively identified as Edwardsiella tarda by MALDI-TOF MS and was broadly susceptible to all antibiotic tested including beta-lactams and fluoroquinolones.
Discussion
Edwardsiella tarda is an infrequently isolated member of the Enterobacterales which is most often associated with gastroenteritis. Reports of additional presentations include peritonitis, intra-abdominal abscess, and wound infections are increasing.1 Bacteremia is rare, and can lead to cholangitis, cholecystitis, and liver abscess via hematogenous spread. Patients of advanced age (>65 years) and those with hepatobiliary diseases including liver cirrhosis and alcohol abuse and iron storage disorders are at increased risk for extraintestinal disease..2 Colonization of the gastrointestinal tract is believed to be a precipitating even leading to bacteremia. Cases of gastroenteritis are usually self-limiting, although ciprofloxacin or trimethoprim/sulfamethoxazole can be utilized in cases of prolonged duration.5 Prompt and accurate diagnosis of E. tarda bacteremia is important as this presentation is associated with an elevated mortality rate,4 particularly among those with liver disease.
As members of the Enterobacterales, Edwardsiella sp. share common biochemical features of other members of the order including being catalase-positive and oxidase-negative. Oxidase negativity aids in the distinction between E. tarda and Aeromonas/Plesiomonas, both species which are also associated with aquatic environments and cause gastrointestinal infections.5E. tarda is non-lactose fermenting and reduces sulfur containing amino acids to hydrogen sulfide. This can result in confusion with members of the genus Salmonella4 as colonies exhibit similar appearances on medias including Hektoen Enteric agar and Xylose-lysine deoxycholate agar formulated for recovery and presumptive identification of Salmonella from gastrointestinal specimens. Importantly, E. tarda is also indole-positive which will biochemically differentiate these two genera.
E. tarda is associated with freshwater or brackish aquatic environments. This organism is a well-known pathogen in aquaculture industries causing serious infections in fish and significant economic loss. As such, vaccines and prophylactic antibiotics are utilized to prevent E. tarda infection among aquatic animals, and diagnostic approaches including specialized multiplex PCRs are available. Human infection is often associated with consumption of contaminated fish and seafood, and diagnosis is almost exclusively dependent on microbiological culture by contrast. Clinical multiplex molecular panels for both gastrointestinal and bloodstream infections lack the ability to detect E. tarda. In this patient’s case, recent consumption of seafood serves as the most likely event leading to E. tarda gastrointestinal colonization and is consistent with previously reported cases of E. tarda bacteremia.3 The patient was treated with meropenem eventually narrowed to piperacillin/tazobactam and blood cultures cleared. He was weaned off pressors and extubated. Unfortunately, the patient decompensated over the course of three weeks due to worsening shock and acidosis. He was moved to comfort care and expired soon thereafter.
References
Hirai et. al. 2015. Edwardsiella tarda bacteremia. A rare but fatal water- and foodborne infection: Review of the literature and clinical cases from a single centre. Can. J. Infect. Dis. Med. Microbiol. 26(6): 313-318.
Hasegawa M., and Sanmoto, Y. 2024. Recurrent cholangitis and bacteremia due to Edwardsiella tarda: a case report. Oxford Med. Case Rep. Volume 2024, Issue 1, January 2024, omad148, https://doi.org/10.1093/omcr/omad148
An et. al. 2023. Case Report: Disseminated Edwardsiella tarda infection in an immunocompromised patient. Front. Cell. Infect. Microbiol. 20 November 2023.
Janda, J.M, and Abbott, S.L. 1993. Infections Associated with the genus Edwardsiella: the Role of Edwardsiella tarda in human disease. Clin. Infect. Dis. Oct;17(4):742-8.
Janda, J.M and Abbott, S.L. 1999. Unusual Food-Borne Pathogens: Listeria monocytogenes, Aeromonas, Plesiomonas, and Edwardsiella species. Clin. Lab. Med. 19(3):553-582.
-Andrew Clark, PhD, D(ABMM) is an Assistant Professor at UT Southwestern Medical Center in the Department of Pathology, and Associate Director of the Clements University Hospital microbiology laboratory. He completed a CPEP-accredited postdoctoral fellowship in Medical and Public Health Microbiology at National Institutes of Health, and is interested in antimicrobial susceptibility and anaerobe pathophysiology.
-Francesca Lee, MD, is an associate professor in the Departments of Pathology and Internal Medicine (Infectious Diseases) at UT Southwestern Medical Center
A 72 year-old male with severe aortic stenosis resulting in heart failure underwent aortic valve replacement and became febrile with mild shortness of breath on post-operative day one. Additional pertinent medical history includes end-stage renal disease secondary to diabetic nephropathy with kidney transplantation seven months prior which was complicated by delayed graft function requiring hemodialysis and immunosuppression. The patient’s presentation was suspicious for either a post-operative fever or a complication of hemodialysis. Physical examination did not suggest infection at either the surgical site or his arteriovenous fistula. A chest CT revealed a moderate, loculated left pleural effusion with left lower lobe atelectasis and nodular consolidations in the right lung, raising concern for pneumonia. A bronchial biopsy was obtained, and histopathological evaluation was consistent with an abscess, but no organisms were visualized. Culture from a left bronchial brushing was also obtained at the same time. Two days later, the patient developed a progressively enlarging forehead lesion (Image 1).
Image 1. Progressively enlarging forehead lesion. Microscopic evaluation following biopsy of the lesion revealed organisms morphologically consistent with Nocardia species.
Laboratory Identification
Microscopic evaluation of the biopsy of the lesion for culture revealed branching, beaded gram positive filamentous bacteria which stained positive using a modified acid-fast stain, suggestive of Nocardia species. This was corroborated by the bronchial culture, where similar modified acid-fast organisms were visualized and grew chalky white colonies identified as Nocardia sp. by MALDI-TOF MS (Image 2). The isolate was referred to a reference laboratory for definitive speciation and antimicrobial susceptibility testing, and the patient was started empirically on ceftriaxone and trimethoprim/sulfamethoxazole. Speciation and susceptibility testing subsequently identified the organism as Nocardia brasiliensis with susceptibility to both ceftriaxone and trimethoprim/sulfamethoxazole, and treatment was thereafter continued for 40 days.
Image 2. Modified acid-fast stain of a bronchial culture from the patient showed a branching, modified acid-fast rods consistent with Nocardia. Cultures of both the lesion and bronchial brushing revealed white, chalky colonies on buffered charcoal yeast extract agar consistent with Nocardia sp.
Discussion
Nocardia is a saprophytic bacterium that is commonly found in soil worldwide. Gram staining of Nocardia sp. usually reveals delicate, weakly or erratically staining, beaded, branching Gram-positive rods. There are currently 130 known species of Nocardia,1 of which more than 50 have been reported to have clinical relevance.2 Disease due to Nocardia is known to occur due to inhalation of the organism from the environment or traumatic inoculation. It is thought to be an opportunistic pathogen most commonly affecting immunocompromised patients3 like the patient in this case.
Manifestations of nocardiosis vary widely depending on the species responsible, with most common presentations being lymphocutaneous, pulmonary, or disseminated disease. Here, we present a patient who displayed both lymphocutaneous and pulmonary forms of Nocardia infection. While prognosis is poorer in immunocompromised patients, with appropriate treatment, recovery rate is high in both lymphocutaneous and pulmonary forms, unlike in disseminated disease, where studies have found mortality rates between 44% to 85%.4
Diagnosis of Nocardia infections is mainly performed through either direct visualization of the organism or through culture. The modified acid-fast stain is commonly performed but has limited sensitivity and should generally be performed in conjunction with the Gram stain.5 Recovery in culture is most reliable from respiratory and/or tissue samples, while blood culture, by contrast, has poorer yield. While some strains, as in this patient, appear within several days in culture, some strains may take up to 2 to 3 weeks to detect5 necessitating extended incubation and consultation with the clinical laboratory.
Treatment requires prolonged courses of antibiotics. Susceptibilities vary by species, making it important to obtain species identification to identify appropriate therapy to guide empiric therapy. Susceptibility testing is performed to allow further tailoring of antibiotic regimens. In this case, the patient was treated with a combination of trimethoprim/sulfamethoxazole and ceftriaxone, both of which cover the majority of clinically relevant Nocardia species, until susceptibility testing revealed that the empiric treatment provided adequate coverage for the Nocardia brasiliensis identified, supporting continuation of the chosen empiric regimen.
Hamdi AM, Fida M, Deml SM, Abu Saleh OM, Wengenack NL. Retrospective Analysis of Antimicrobial Susceptibility Profiles of Nocardia Species from a Tertiary Hospital and Reference Laboratory, 2011 to 2017. Antimicrob Agents Chemother. 2020 Feb 21;64(3):e01868-19. doi:10.1128/AAC.01868-19.
McNeil MM, Brown JM. The medically important aerobic actinomycetes: epidemiology and microbiology. Clin Microbiol Rev. 1994 Jul;7(3):357-417. doi:10.1128/CMR.7.3.357.
Traxler RM, Bell ME, Lasker B, Headd B, Shieh WJ, McQuiston JR. Updated Review on Nocardia Species: 2006-2021. Clin Microbiol Rev. 2022 Dec 21;35(4):e0002721. doi: 10.1128/cmr.00027-21.
Saubolle MA, Sussland D. Nocardiosis: review of clinical and laboratory experience. J Clin Microbiol. 2003 Oct;41(10):4497-501. doi: 10.1128/JCM.41.10.4497-4501.2003.
-Albert Budhipramono, MD, PhD, is currently a PGY1 Clinical Pathology Resident at the University of Texas Southwestern Medical Center in Dallas, Texas.
-Clare McCormick-Baw, MD, PhD is an Assistant Professor of Clinical Microbiology at UT Southwestern in Dallas, Texas. She has a passion for teaching about laboratory medicine in general and the best uses of the microbiology lab in particular.
-Andrew Clark, PhD, D(ABMM) is an Assistant Professor at UT Southwestern Medical Center in the Department of Pathology, and Associate Director of the Clements University Hospital microbiology laboratory. He completed a CPEP-accredited postdoctoral fellowship in Medical and Public Health Microbiology at National Institutes of Health, and is interested in antimicrobial susceptibility and anaerobe pathophysiology.
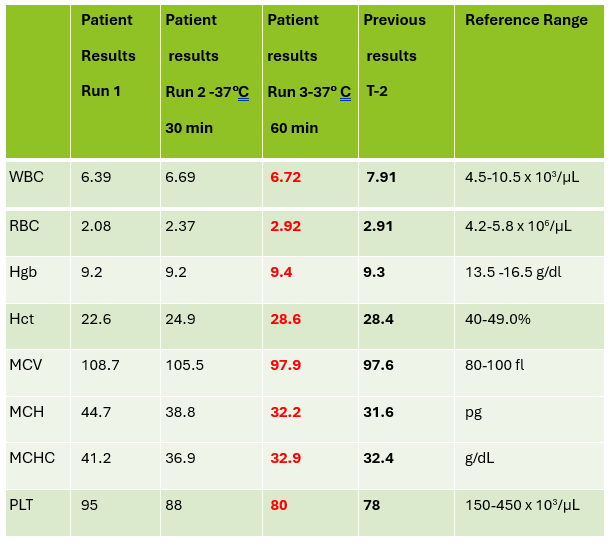
A CBC was received on a 70-year-old surgical inpatient at our facility and was run in automated mode on our Sysmex XN analyzer. On the first run, the analyzer gave flags for RBC agglutination and MCHC >37.5. These flags require evaluation of the high MCHC with investigation of a cold agglutinin, lipemia or icterus. A smear review for RBC agglutination was also indicated. The sample was incubated at 37°C for 30 minutes and the CBC was repeated. Results are shown below in Table 1.
Table 1. CBC results before warming at 37°C and after 15 minutes incubation.
On Run 2, the MCHC is now below 37.5, so there was no operator alert to incubate further or to investigate the high MCHC. This result was validated with the comment “37°C results, possible cold agglutinin.” However, there was still an RBC agglutination flag, a high MCV, and the hemoglobin and hematocrit still don’t look great, i.e. they don’t follow ‘the rules of 3’. We know that these rules are only valid for normal samples, but if we look at the previous results from 2 days ago, the Hgb 9.3 and Hct 28.4 did follow the ‘rules of 3’ and additionally, today’s results look like the patient’s hemoglobin has not changed but the hematocrit has dropped. The RBC did go up on the second run, but it is considerably lower than 2 days ago. The indicies are also inconsistent with the previous sample. “Hmmm…What could cause this?” Instead of validating, this is what you should be asking yourself.
Cold agglutinins are known for causing pre-analytical and analytical spurious results with CBCs. With cold agglutinins, IgM antibodies bind to the RBCs after exposure to cold, causing RBC agglutination which leads to a classic pattern of false results. There is an increased MCV because the RBCs are clumped and sticking to one another, making the analyzer think these larger clumps are individual RBCs. This, in turn, will make the RBC count appear deceased because large clumps of RBCs going through the RBC aperture are counted as one RBC. The hematocrit is lowered because the volume of a clump is less than those cells individually. Hemoglobin concentration, on the other hand, is not affected by cold agglutinins, but because the hematocrit and RBC are falsely lowered, this makes the MCH and MCHC spuriously markedly increased. Cold agglutinins may be subtle, like this one, but some have extremely high MCHC and MCV, and extremely low RBC and Hct. Clues that you have a cold agglutinin are not only the high MCHC, but also flags from your analyzer such as “RBC agglutination”. The hematocrit will likely seem too low for the hemoglobin, and you may even see a hemoglobin that is higher than the hematocrit (yes it happens!) and RBC values so low as to be incompatible with life.
The first thing is to do if you get these spurious results is to compare the parameters and instrument flags and decide if the results are consistent with a cold agglutinin. Other factors that may cause a high MCHC include lipemia, icterus, low sodium, abnormal proteins, hemolyzed samples and samples from patients with hemoglobinopathies. The patterns of result with these samples will show a high MCHC, but with a normal or low MCV, and you won’t usually see an RBC agglutination flag. A sample with cold agglutinins may also appear grainy or clumpy to the naked eye. With a suspected cold agglutinin, warming the sample for 15-30 minutes will allow the RBCs to disperse and improve the results. However, these results must be reviewed, and if there are still instrument flags and/or if there is still clumping, the results may not yet be corrected or ‘correct’. Be sure to review a smear from the tube before warming and after. The first smear confirms the presence of the cold agglutinins and clumping, and the second smear should confirm the resolution of the clumping after warming.
This sample did have the characteristic grainy appearance of a cold agglutinin, and the post warming results did look a bit better, but the RBC agglutination flag was still present, and a review of the smear showed that the sample still had RBC clumping. After warming for another 30 min, the sample was quickly mixed and placed back on the analyzer. The results of this 3rd run are shown below, in Table 2.
Figure 1. EDTA tube with RBC agglutinationTable 2. CBC results on 3 runs. Run 3, after 60 min of incubation.
Notice that hemoglobin has not changed as it is not affected by the cold agglutinins, but after warming for 60 minutes, the RBC, hematocrit and indicies all now look consistent with the previous sample drawn 2 days ago, and a review of the smear showed no RBC agglutination. These results from the 3rd run are ready to validate.
Cold agglutinins are IgM autoantibodies that react best at 4°C but may also react at room temperature. They are generally not clinically significant and may be found in many healthy individuals. These natural cold autoantibodies occur at low titers, less than 1:64, and have no activity at higher temperatures. However, because they react at room temperature, they are notorious as a pre-analytical and analytical factor that causes spurious CBC results. They can also cause difficulties in Blood Banking during ABO/Rh typing and antibody detection.
Cold agglutinins have various clinical manifestations. Benign cold agglutinins generally do not cause hemolytic anemia and need no treatment. Most benign cold autoantibodies have anti-I specificity, are polyclonal, low titer, and do not react above 30°C. Cold agglutinins associated with Mycoplasma pneumoniae, and infectious mononucleosis are usually clinically insignificant. In cases where they do cause hemolytic anemia, the antibodies are polyclonal IgM with normal κ and λ light chains. The anemia is acute and generally spontaneously resolves in several weeks without treatment.
Though most cold agglutinins are benign and do not cause RBC destruction, when they do, they can cause hemolytic anemia that varies in severity from mild to life-threatening. This chronic cold agglutinin disease (CAD) is now known to be a form of autoimmune hemolytic anemia caused by a bone marrow lymphoproliferative disorder. Chronic CAD is a cold-autoantibody autoimmune hemolytic anemia (cAIHA) that is caused by an autoantibody produced by the clonal B cell lymphocytes. This antibody is usually monoclonal IgM with κ light chains and “I” or “i” specificity. These pathological cold agglutinins are high titer and usually react at 28°C to 32°C, and even up to 37°C. The highest temperature at which the antibodies continue to be activated is called the thermal amplitude. Because these can act at higher thermal amplitude, they may lead to CAD. In CAD the IgM autoantibodies bind to red cell antigens at 30-32°C, typically in the cooler extremities. IgM’s structure, a large immunoglobin pentamer, makes it an effective activator of the classical complement system. As the blood circulates to the central parts of the body, the RBCs warm up and the IgM antibodies dissociate from the RBC membranes, but the complement activation will continue, leading to RBC hemolysis and a cAIHA.
Chronic CAD occurs most often in adults over 50, is more common in women, and produces anemia with varying severity. Patients may be seasonally affected. In the winter, the temperature of blood may fall below 30°C in the extremities, activating the cold agglutinins. Patients may experience acrocyanosis of the hands, feet, ears, and nose with exposure to cold. They may also experience other cold related symptoms such as numbness and Raynaud’s. Patients with chronic CAD and mild anemia are therefore monitored with a ‘wait and see’ plan and advised to avoid cold temperatures. In patients with more severe anemia, it is found that targeting the underlying lymphoproliferative disorder provides the best treatment. Rituximab has been used to achieve partial remission. Therapeutic plasma exchange is also used in severe cases to rapidly remove cold agglutinins.
I have been thinking about cold agglutinins recently because of the number I have seen come into our lab this winter. As I am writing this, watching the temperature outside drop in anticipation of more snow coming in tonight, cold agglutinins came to my mind again. I used to live in the cold northern Northeast, but after moving further south, we see fewer cold agglutinins in hematology than I used to see. This winter we have had some cold spells, and, interestingly, I’ve seen more cold agglutinins. That led me to ask myself if cold agglutinin disease is really more common when patients are exposed to cold temperatures. I remember learning myself, and telling my students that that the treatment for mild to moderate CAD was to advise patients to move to someplace warmer. It has been assumed for many years that CAD worsens in colder climates or seasons. Interestingly, there have been a number of studies done since the 1950’s that examined the relationship of cold temperatures and CAD. The studies used hemoglobin, bilirubin and LDH for monitoring. Early case reports had findings that supported the theory of more anemia and higher LDH in the winter. (Dacie, Lyckholm)One recent article in 2022 found a 4-fold difference in the incidence of CAD between cold (Norway) and warm (Italy) climates. (Berentsen). However, atthe same time another study found that there was no statistically significant seasonal variation in hemoglobin, but that LDH levels were higher in winter. It concluded that these conditions should be monitored through all seasons because of the risk of hemolysis and thrombotic episodes. It was also demonstrated that though there may not be obvious statistical difference in CAD between cold and warm months, that there is a large variability of disease severity across patients and even with an individual patient. (Roth).
In conclusion, when working in any department of the laboratory, quality results are important. Results on the patient chart that vary considerably from day to day because sometimes a cold agglutinin has been effectively resolved in lab testing and other days results after 15-30 minutes of warming are just reported without a good review of the smear and the parameters, are confusing, and could affect patient care. If one tech reports the results after 30 min incubation with values that are still spurious, and the next tech resolves the agglutination with further warming, the lab will be reporting out inconsistent results. A patient who actually has stable CBC results may have deltas and what appear to be erratic results. Cold agglutinins do take time to resolve, but with over 80% of samples autoverifying with the use of auto verification, we have time to work on these problem samples. If something doesn’t look or feel right about a sample, look at all the parameters, check the instrument flags and operator alerts, check the previous results and investigate any changes. It is important to review results carefully, because we want to report out the best results possible.
References
S Berentsen, W Barcellini, S D’Sa, U Randen, THA Tvedt, B Fattizzo, E Haukås, M Kell…
Blood, The Journal of the American Society of Hematology, 2020•ashpublications.org
Climent F, Cid J, Sureda A. Cold Agglutinin Disease: A Distinct Clonal B-Cell Lymphoproliferative Disorder of the Bone Marrow. Hemato. 2022; 3(1):163-173. https://doi.org/10.3390/hemato3010014
Nikousefat Z, Javdani M, Hashemnia M, Haratyan A, Jalili A. Cold Agglutinin Disease; A Laboratory Challenge. Iran Red Crescent Med J. 2015 Oct 17;17(10):e18954. doi: 10.5812/ircmj.18954. PMID: 26566452; PMCID: PMC4636857.
Patriquin, C.J. and Pavenski, K. (2022), O, wind, if winter comes … will symptoms be far behind?. Transfusion, 62: 2-10. https://doi.org/10.1111/trf.16765
Rodak, Bernadette F., et al. Hematology: Clinical Principles and Applications. 5th ed. St. Louis, Mo., Elsevier Saunders, 2016
Röth A, Fryzek J, Jiang X, Reichert H, Patel P, Su J, et al. Complement-mediated hemolysis persists year round in patients with cold agglutinin disease. Transfusion. 2022; 62: 51–59. https://doi.org/10.1111/trf.16745
-Becky Socha, MS, MLS(ASCP)CMBBCM graduated from Merrimack College in N. Andover, Massachusetts with a BS in Medical Technology and completed her MS in Clinical Laboratory Sciences at the University of Massachusetts, Lowell. She has worked as a Medical Technologist for over 40 years and has taught as an adjunct faculty member at Merrimack College, UMass Lowell and Stevenson University for over 20 years. She has worked in all areas of the clinical laboratory, but has a special interest in Hematology and Blood Banking. She currently works at Mercy Medical Center in Baltimore, Md. When she’s not busy being a mad scientist, she can be found outside riding her bicycle.
In the 1920s, the Greek physician Georgios Papanikolaou developed a method of cervical cancer screening, now reliably used and colloquially known as a “Pap smear”. During a Pap smear, a healthcare provider swabs cells from the cervix for further analysis by the lab1, including cytopathologic examination. Regular utility of Pap smears in women aged 21 to 65 has decreased the incidence and mortality of cervical cancer by at least 80% in the United States alone2. 95% of cervical cancer cases worldwide are caused by persistent human papilloma virus (HPV) infection of the cervix3. However, other sexually transmitted infections (STIs) and common non-STIs with associated morphological changes can also be identified on Pap smears.
Bacteria
Bacterial vaginosis (BV) is the most common cause of abnormal discharge in young women, characteristically with a “fishy” ammonia-like odor. Although not strictly considered an STI, this infection is associated with severe rare complications including pelvic inflammatory disease (PID), infertility and premature labor. The vagina typically have an abundance of Lactobacillus spp., which is a gram positive bacilli (Figure 1). However, if the normal vaginal flora is disrupted with high abundance of gram negative bacteria such as Gardnerella vaginalis or Mobiluncus, then BV may occur. On pap smears, ‘clue cells’ which are squamous epithelial cells with coccobacilli, appear as dark purple staining and cloudy appearance suggest BV. In cervicovaginal samples typically found in women using intrauterine device usage, clumps of filamentous bacteria suggestive of Actinomyces species may also be visible. For cases pertaining to bacterial etiologies, staining of the rods or filaments usually warrant suspicion of infection. Other the other hand although Chlamydia trachomatis is the most common bacterial STI in the United States, with up to 4 million new cases every year, diagnosis on pap smears is not the definitive diagnosis since the cytopathology is non-specific. The interpretations include visualization of inflammatory exudates and inflammatory cells. Oftentimes, further testing is needed.
Figure 1. Lactobacilli spp. bacilli seen as part of normal flora (red arrow, top left and top middle). Example of a clue cell (red arrow): intermediate squamous cell coated with grape-like short coccobacilli (Gardnerella vaginalis) with a shift in normal flora (top right). What appears to be a clue cell (red arrow) at first glance is actually an intermediate squamous cell covered with normal flora upon closer inspection (bottom left). Follicular cervicitis due to Chlamydia infection (bottom right). Note lymphohistiocytic aggregates with polymorphous lymphocytes and histiocytes (red arrow). Tangible body macrophages may also be identified (ThinPrep, 40X).
Parasite
Trichomonasvaginalis is the most prevalent non-viral STI in the United States, strongly associated with an increased risk of HIV infection among women4. Majority of the patients may exhibit symptoms such as burning, itching and vaginal discharge. T. vaginalis is a parasitic flagellated protozoan although in some cases, a flagella may be found. Typically, it is of a pear-shape and staining of a nucleus may be visualized (Figure 2).
Figure 2. Pear-shaped round-oval Trichomonas spp. (red arrows) ranging in size from 15-30 µm present singly and in groups, known as “Trich parties”. Note pale vesicular eccentrically located nucleus and eosinophilic cytoplasmic granules. Reactive cytological changes (black arrow) such as nuclear enlargement and perinuclear halos are also identified. (ThinPrep, 40X. Case courtesy of Dr. Edina Paal, VA Medical Center, Washington DC).
Fungus
Yeast infections (vulvovaginal candidiasis) due to Candida infection occur in 75% of women at some point in their lives. Classic clinical presentation includes itching, erythema, and thick white “cottage-cheese” discharge. There is also an increased association with HIV infection, diabetes, and any cause of immunosuppression (e.g. transplant, chemotherapy, steroids). Morphologically, budding yeast forms (conidia, small and oval measuring 3-6 µm) and pseudohyphae (long filamentous spores) are found. The combination of pseudohyphae and yeast forms are referred to as “sticks and stones” and frequently, squamous cells lined up along the pseudohyphae are found referred to as having “shish kebab” appearance (Figure 3)5. Note that no true septation is found.
On pap smears, sometimes there could be superficial mucosal infections that may appear associated with enlarged hyperchromatic nuclei with halos, which can be confused with low grade squamous intraepithelial lesions.
Figure 3. Shish kebab: entangled streaming intermediate and superficial squamous cells along Candida spp. pseudohyphae (red arrow, top). Partially treated Candida infection. Note poorly formed granular pseudohyphae (red arrow) with significant treatment effect (red arrow, bottom) (ThinPrep, 40X).
Viruses
HPV with its associated risk of cervical cancer remains the most crucial microorganism to detect on Pap smears. Given HPV’s association to cancer development, it is crucial to examine the samples for cervical lesions and their associated pathologies. HPVs in the low-risk category typically is associated with low-grade squamous intraepithelial lesions and HPVs in the high-risk category is associated with high-grade squamous intraepithelial lesions and invasive squamous cell carcinoma. Other viral etiologies seen in PAP smears include Herpes simplex virus (HSV) and cytomegalovirus (CMV). Infections with HSV can be asymptomatic but the most common symptom is the development of vesiculopustular or small ulcerative lesions on the genitalia. The classic cytopathological findings of HSV infection are the 3 “Ms”: multinucleation, nuclear molding, and margination of chromatin. CMV infections are rare, usually asymptomatic and can be transient. On the pap smears, the infected cells may appear with intranuclear inclusions surrounded by a halo.
Conclusions
While Pap smears are routinely performed on women and can provide a presumptive diagnosis, current developments in molecular technologies (e.g. Nucleic acid amplification tests (NAATs)) is transforming the field. There are several FDA-approved molecular platforms available for clinical diagnostic labs to test for HPV and can even genotype the strain to determine risk levels. Crucially, the remaining sample from liquid-based Pap tests6 can also be submitted for both NAAT testing and HPV-DNA testing, facilitating a quicker turnaround time and obviating the need for additional patient sampling. Most recently, a PCR-based test is now on the market (Cepheid, Sunnyvale, CA) that can diagnose all three etiologies, BV, Candidiasis, and Trichomoniasis, within 60 minutes from a single specimen. In summary, accurate examination of the Pap smear often incidentally provides the first step in the diagnosis and further work-up of all these infectious diseases.
3. Lei J, Ploner A, Elfström KM, Wang J, Roth A, Fang F, et al. HPV Vaccination and the Risk of Invasive Cervical Cancer. New England Journal of Medicine. 2020 Oct 1;383(14):1340–8.
4. Davis A, Dasgupta A, Goddard-Eckrich D, El-Bassel N. Trichomonas vaginalis and Human Immunodeficiency Virus Coinfection Among Women Under Community Supervision: A Call for Expanded T. vaginalis Screening. Sex Transm Dis [Internet]. 2016 Sep 15 [cited 2024 Feb 5];43(10):617–22. Available from: https://pubmed.ncbi.nlm.nih.gov/27631355/
5. Kamal Meherbano M. The Pap smear in inflammation and repair. Cytojournal [Internet]. 2022 Apr 30; 19:29. Available from: doi: 10.25259/CMAS_03_08_2021
6. Hawthorne CM, Farber PJ, Bibbo M. Chlamydia/gonorrhea combo and HR HPV DNA testing in liquid-based pap. Diagn Cytopathol [Internet]. 2005 Sep [cited 2024 Feb 5];33(3):177–80. Available from: https://pubmed.ncbi.nlm.nih.gov/16078250/
-Zoon Tariq is a pathology resident at George Washington University. Her interests include surgical pathology and cytopathology.
-Rebecca Yee, PhD, D(ABMM), M(ASCP)CM is the Chief of Microbiology, Director of Clinical Microbiology and Molecular Microbiology Laboratory at the George Washington University Hospital. Her interests include bacteriology, antimicrobial resistance, and development of infectious disease diagnostics.