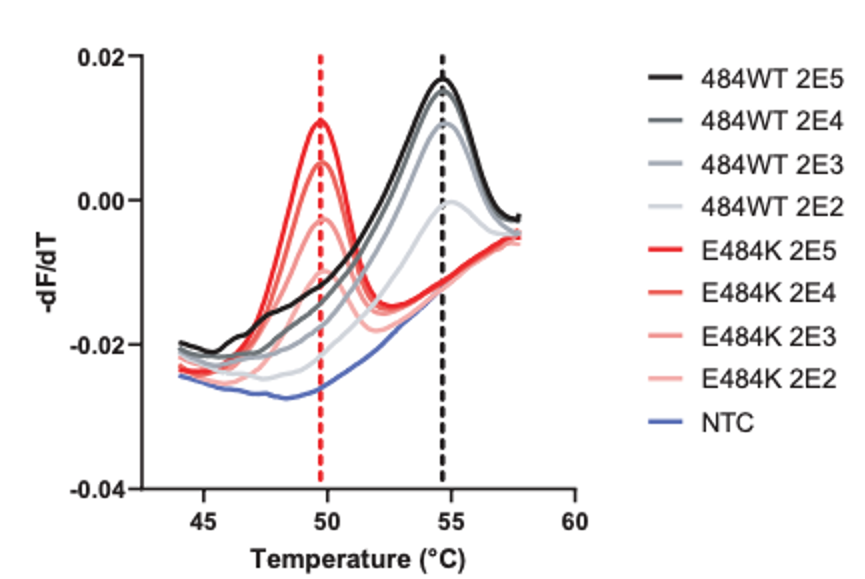
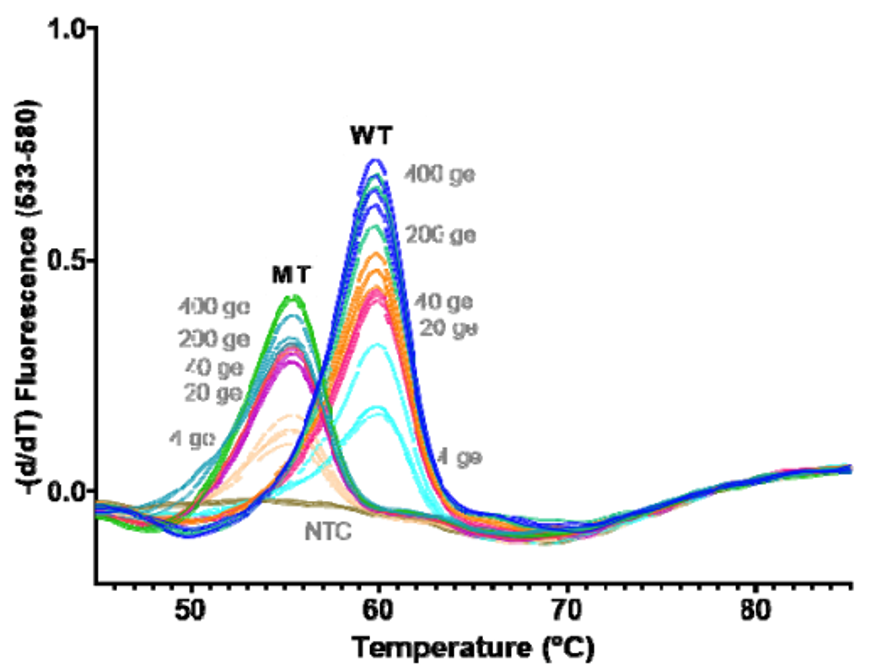
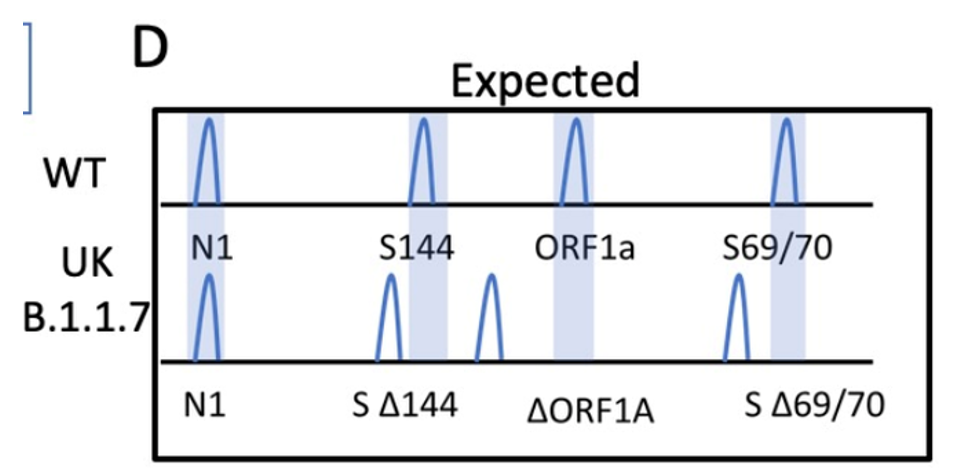
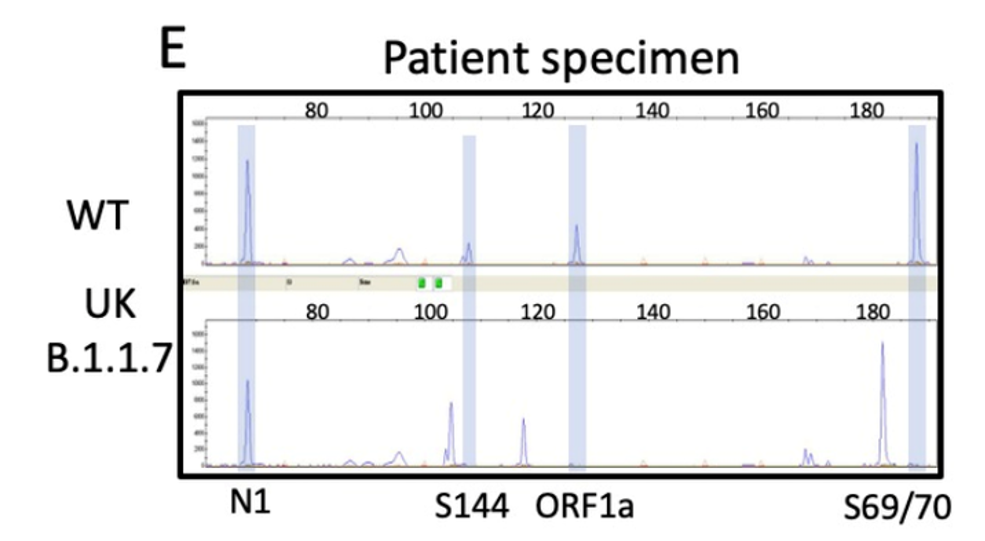
While you have surely heard about all of the SARS-CoV-2 variants and how concerning they are, I would bet that you may not know how they are classified. Sure, from my last post, the technical aspects of whole genome sequencing and targeted approaches have been described, but bioinformatic (big data) analyses are essential to assign lineages. Furthermore, the advances of machine learning have been integrated into this system for SARS-CoV-2 lineage assignment.
How VOC lineages are given
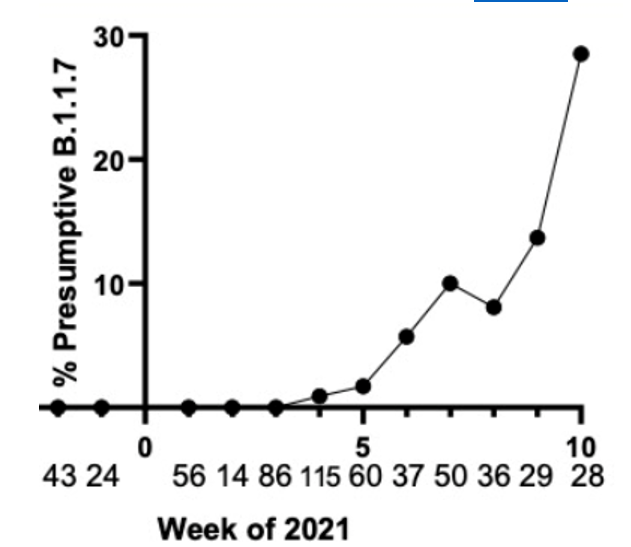
First, phylogenetic trees (circular example below) are formed to demonstrate relatedness of strains based on how many mutations they share. The more similar they are, the closer they are together. These trees are not new nor do they rely on artificial intelligence, but they can give visual clues as to whether a lineage is new. For instance, when the first variant of concern B.1.1.7 (now called Alpha) was discovered, it branched away from other limbs of the phylogenetic tree.
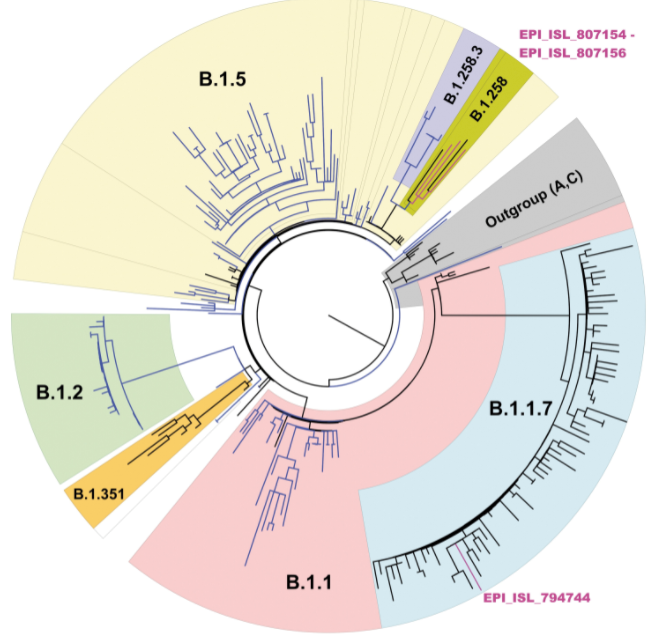
Within these new viral variants, there are a set of mutations that are present in most of the viral variants. For instance, there are 17 protein coding changes in Alpha variant. However, these exact 17 mutations may not be in every Alpha variant. Individually, mutations may be present in 98% of isolates or lower; the spike gene deletion of amino acids 242-244 of the Beta variant (B.1.351, South Africa origin) is only present in 88% of specimens sequenced. This could be due to issues in sequencing, data processing, or just the prevalance/biology of the virus.
As there are many mutations that fit into certain variants, it would be difficult for a human to process all of this information in a probabilistic manner to assign lineages. Thus, machine learning tools (most common SARS-CoV-2 program is Pangolin) have been added onto the end of bioinformatic analyses to assign the lineage to a sample.
How machine learning works
The subject of machine learning has been discussed in a previous post about Protein folding prediction. Briefly, it is helpful to remember that machine learning is a process to create algorithms that give an outcome based on training data. The more diverse, large, and well curated the data, the better the accuracy of the program. One pitfall is they are based on previous data, which works well for many situations: using AI to find a lung cancer on chest x-ray would work well, because lung cancers have consistent characteristics.
However, with COVID-19, new variants keep arising and current variants are evolving (think Delta and Delta “plus”). Furthermore, if the classifier Pangolin is trained on high quality data, then trying to interpret lower quality data (missing genome regions, few sequencing reads) may confuse Pangolin and lead to inaccurate results. What follows is an example of how this occurred at our institution.
Case study
We have been sequencing COVID-19 positive specimens at UT Southwestern for the last several months. Many of the cases have been the Alpha variant (B.1.1.7, origin U.K.). However, it was around this time that Delta (B.1.617.2, origin India) cases started to arise. In one week, we found two specimens that were classified as B.1.95. This was an unusual variant I had not heard of before. There are several “wild type” strains that are B.1.1/ B.1.2 and other derivations, but I had not seen anything like this before.
Clinical history
Two specimens sequenced belonging to Hispanic, adolescent brothers whose mother had recently been hospitalized with COVID-19. There was information on mother’s travel history.
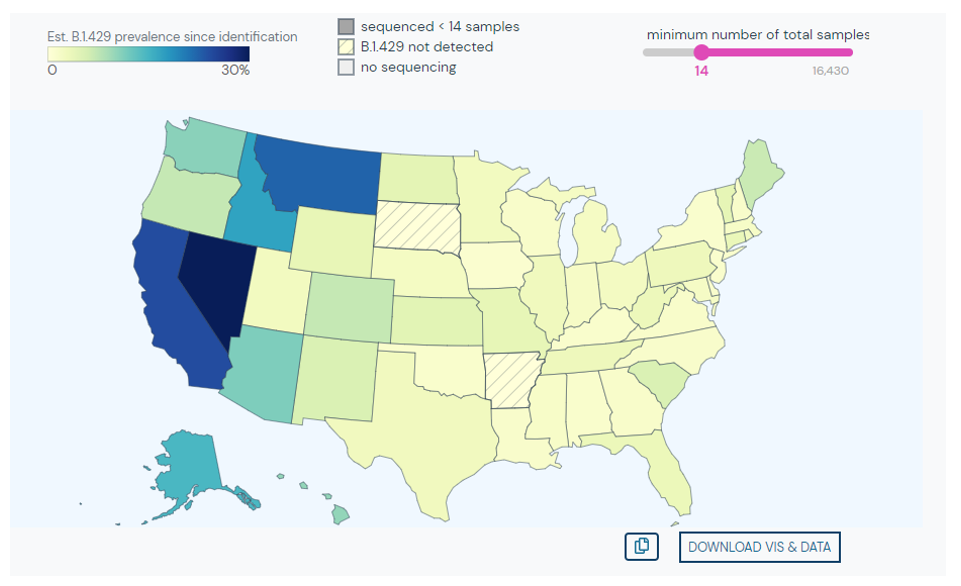
Therefore, I performed manual review of the specific variants. Many of the diverse mutations occur in the spike protein, so this was analyzed first. Immediately, I noticed two classic mutations of the Delta variant: a 2 amino acid deletion in the spike gene (S:Del157_158) and a receptor binding site mutation (S:L452R) also seen in the variant from California (B.1.429). Other mutations could be evaluated, but the combination of these two mutations is unique to Delta variant.
One suspected cause was that the Pangolin lineage classifier had an issue. Specifically, it had not been updated since February 2021- when Delta did not exist. Thus, there was no data for the program to classify the variant properly. Upgrading to the latest version of Pangolin provided the correct lineage classification.
A Few weeks later…
Once again, I was checking the lineages reported by the classifier and there were several B.1.617.2 and B.1.617.1. Both of these are variants from India (before the helpful WHO Delta designation), but they are distinct sub-variants. It was odd to see B.1.617.1, because this was found to be less infectious compared to the dominant B.1.617.2 variant (later named Delta) and B.1.617.1 was not spreading across the globe.
Intervention:
Therefore, I once again went to the sequence data for the spike protein to compare some mutations. Although these are sub-variants from the same original variant, they have several mutually exclusive mutations in the spike protein. The figure below compares the prevalence of specific mutations in the spike protein of B.1.617.1 and B.1.617.2 (dark purple = common in a variant, white = rare).
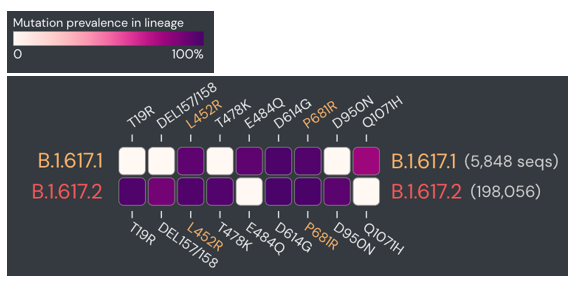
Upon manual review, all of the spike gene mutations were specific to B.1.617.2. So why was there an issue in classification? Again, there were few sequences for either of these sub-variants at that time, so the classifier wasn’t as well trained. Updating the Pangolin version brought the benefit of new data and more accurate classifications.
Take away messages
- Updating Lineage classification software (Pangolin) on a regular basis is needed for accurate results.
- Manual review is essential for any abnormal findings- a typical process for pathologists, but also plays an important role in COVID-19 variant monitoring.
- Know what you’re looking for and know which mutations differentiate the variants.
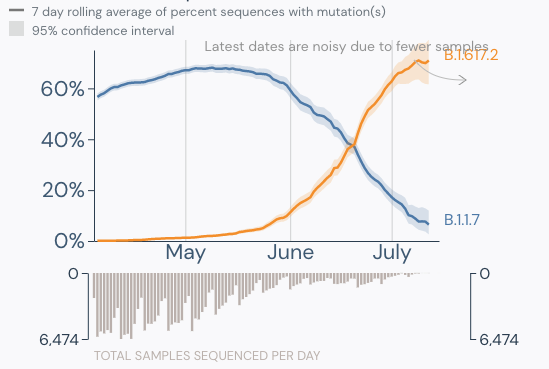
- Delta is now the dominant strain in the U.S. (graphic below).
References
- Outbreak.info
- https://pangolin.cog-uk.io/

–Jeff SoRelle, MD is Assistant Instructor of Pathology at the University of Texas Southwestern Medical Center in Dallas, TX working in the Next Generation Sequencing lab. His clinical research interests include understanding how lab medicine impacts transgender healthcare and improving genetic variant interpretation. Follow him on Twitter @Jeff_SoRelle.