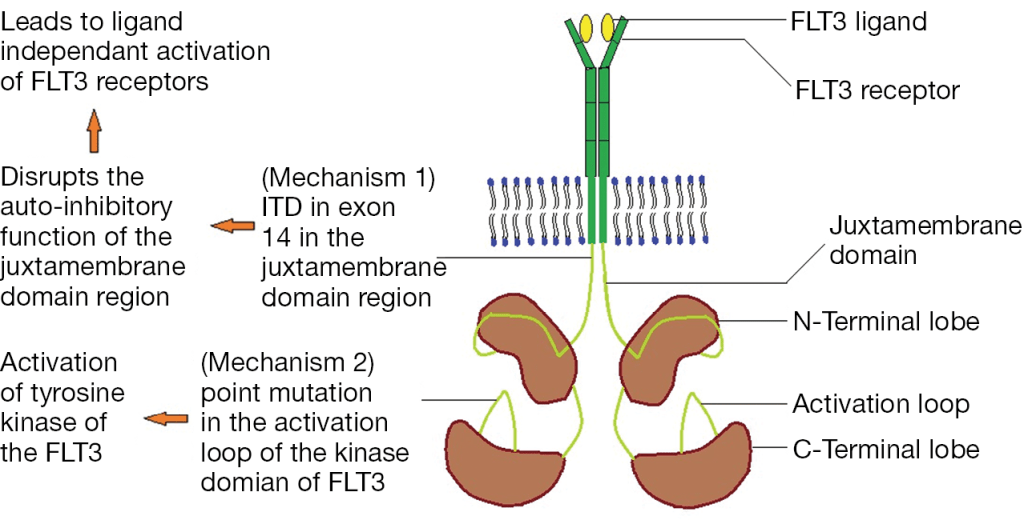
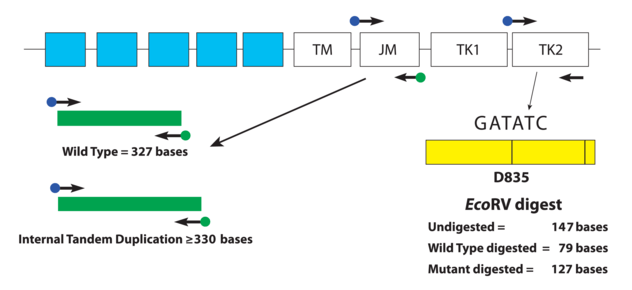
It’s a little deja-vu writing this title one year after a similar blog post on how to validate a COVID-19 assay at the start of the pandemic. In many ways, the challenges are similar: limited reagents/control material, and rising case counts. At least now, there is increasing support in the way of funding from the federal government that could help with monitoring and surveillance. I’m going to summarize the current methods available for detecting the Variants of Concern and emerging variants.
Whole Genome Sequencing
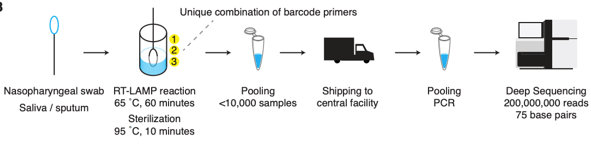
The principle method used by many is whole genome sequencing. It has the advantage of being able to comprehensively examine every letter (nucleotide) of the SARS-CoV-2 genome (30 kilobases long). At our institution, I’ve been working on the effort to sequence all of our positive specimens. While it is achievable, it is not simple nor feasible at most locations. Limitations include:
- Financial: must already own expensive sequencers
- Expertise: advanced molecular diagnostics personnel needed who perform NGS testing
- Data Analytics: bioinformatics personnel needed to create pipelines, analyze data and report it in a digestible format.
- Timing: the process usually takes a week at best and several weeks if there is a backlog or not enough samples for a sequencing run to be financially viable.
- Sensitivity: the limit of detection for NGS is 30 CT cycles, which for us includes only about 1/2- 1/3 of all positive COVID19 specimens.
Bottom line: WGS is the best at detecting new/ emerging strains or mutations when cost/ time is not a concern.
Mutation Screening
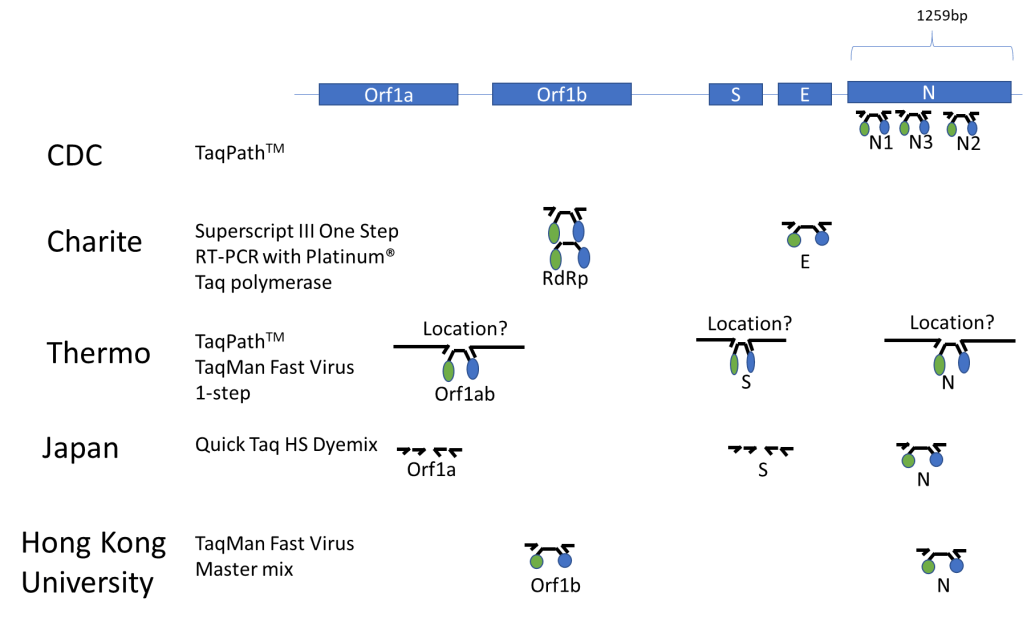
Other institutions have begun efforts to screen for variants of concern by detecting characteristic mutations. For instance, the N501Y mutation in the spike protein is common to the major Variants of Concern (UK B.1.1.7, Brazil P.1, and S Africa B.1.351) and E484K is present in the Brazil (P.1), S Africa (B.1.351) and New York Variant (B.1.526). Thus, several institutions (listed below) took approaches to 1) screen for these mutations and then 2) perform WGS sequentially.
| Institution | Method | Targets |
| Hackensack Meridian Health (HMH) | Molecular Beacon Probes, melting temp | N501Y, E484K molecular beacons |
| Rutgers, New Jersey | Molecular Beacon Probes, melting temp | N501Y molecular beacons |
| Vancouver | Probe + melting curve (VirSNiP SARS-CoV-2 Mutation Assays) | N501Y screen + qPCR reflex; Probe, melt curve assay |
| Yale | RT-qPCR probe assay | S:144del, ORF1Adel |
| Columbia | RT-qPCR probe-assay | N501Y, E484K |
As you can see, HMH, Rutgers and Vancouver are using assays that use probes specific to characteristic alleles combined with melting temperature curves to detect a mutation induced change. Melting curve analysis is normally performed after qPCR to ensure that a single, correct PCR product is formed. This measure is calculated based on the change in fluorescence that occurs when the fluorescent marker is able to bind to its target DNA. Thus the Tm (melting temperature) is similar to the annealing temperature. In this case where a mutation is present in the probe (DNA fragment) binding site, binding is disrupted and occurs at lower a temperature as seen by the downward shift of 5 degrees Celsius in the graph below.
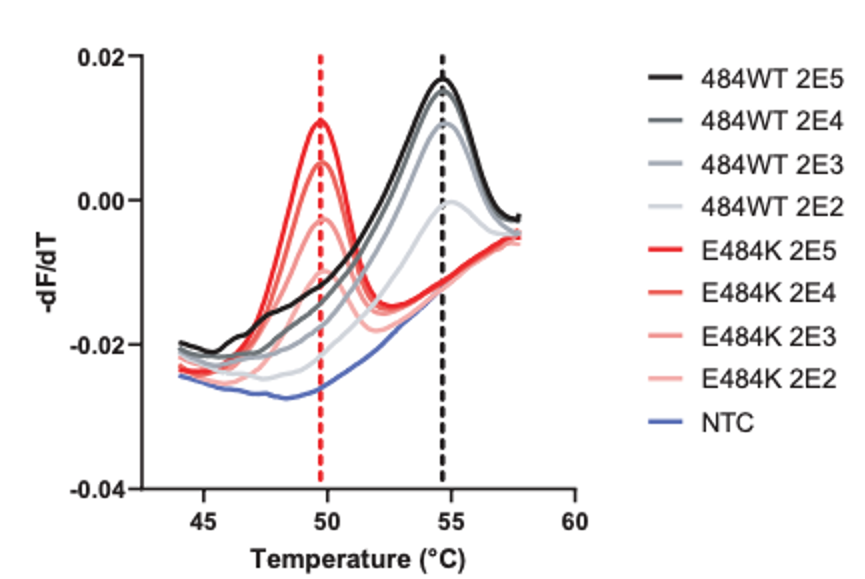
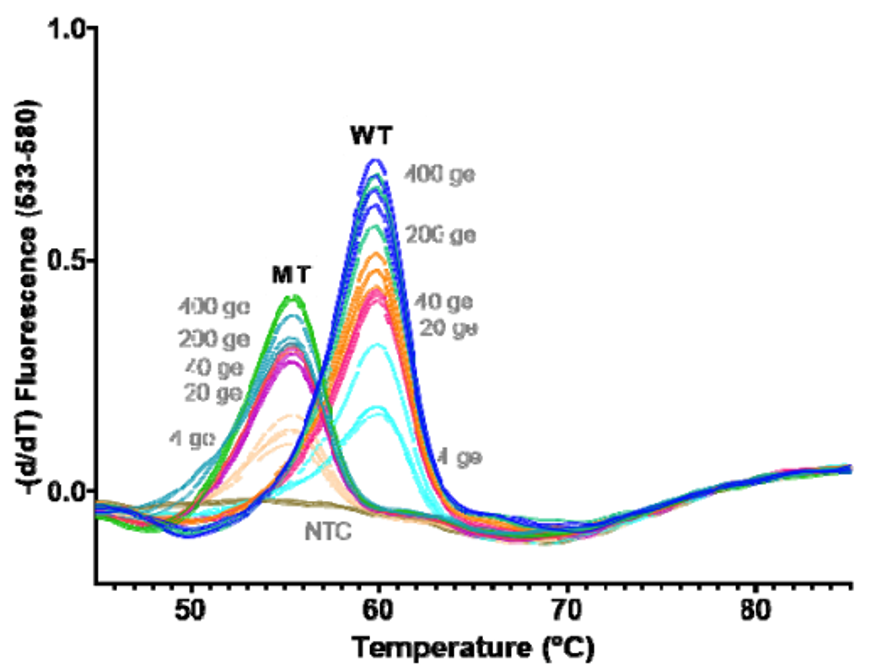
These approaches are quick, but can only perform a 2-3 reactions per well and require much of the same expenses as diagnostic RT-qPCR assays. Most of the studies describe this method as a way of screening for samples to be NGS sequenced, however they will not be as good at detecting emerging strains. For example, the N501Y mutation is not present in the New York nor California variants.
Multiplex RT-qPCR can solve some of these problems. At Columbia and Yale, multiple targets are designed to detect B.1.1.7 (N501Y only at Columbia and S144del + ORF1A del at Yale) vs. Brazil/ S. Africa variants (N501Y & E484K at Columbia and ORF1A only at Yale). As new variants have arrived, we found the New York strain carrying both ORF1A deletion and the E484K mutation. It is now clear there are some hotspot areas for mutation within the SARS-CoV-2 genome, which can complicate interpretations. Therefore, these RT-PCR assays are still useful for screening, but do not replace the need for Whole Genome Sequencing.
Genotyping
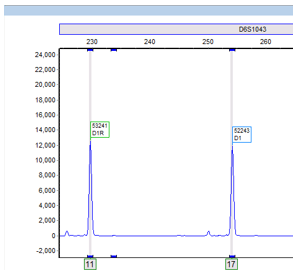
Given the overlapping spectrum of mutations, it would be helpful to test several markers all at once in a single reaction. At a certain point, this would effectively “genotype” a variant as well as WGS. The assays above have been limited to 2 targets/ reaction due to limited light detection channels. Therefore, I’ve created a multiplex assay that can be scaled up to include 30-40 targets within a single reaction without the need for expensive probes. This method is multiplex PCR fragment analysis, which is traditionally used for forensic fingerprinting or bone marrow transplant tracking. In this method, DNA of different length is amplified by PCR, then separated by capillary electrophoresis-the same instrument that performs Sanger Sequencing.
Fragment analysis can be performed to detect deletion/ insertion mutations and single nucleotide polymorphisms (SNPs) by allele-specific primers or with restriction enzymes that only cut the WT or Mutant sequence.
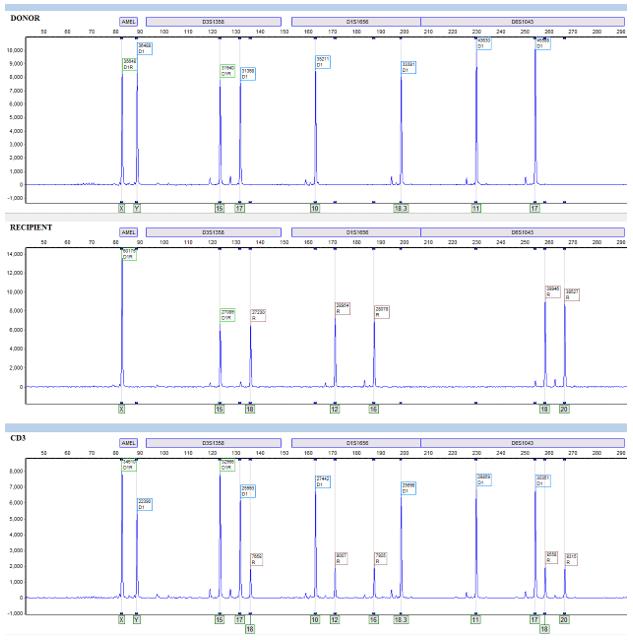
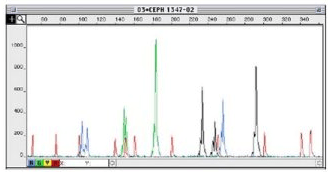
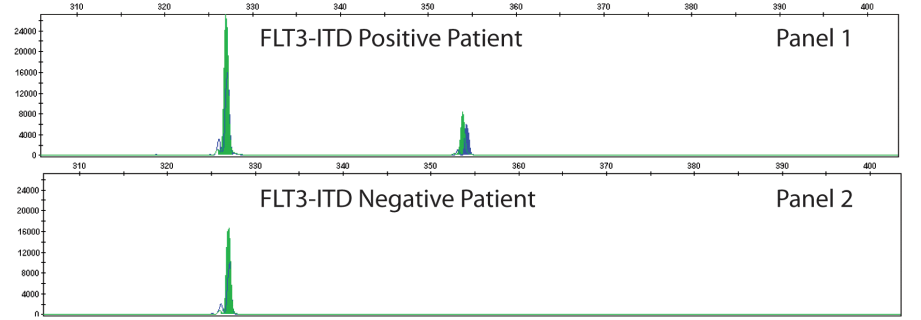
I designed the assay to target 3 deletion mutations in B.1.1.7: S:D69_70, S: D144, and ORF1A: D3675_3677. Each deletion has a specific length and if 3/3 mutations are present, then there is 95% specificity for the B.1.1.7 strain. Samples from December to present were tested and in the first batch, I detected the characteristic B.1.1.7 pattern (expected pattern and observed pattern below).
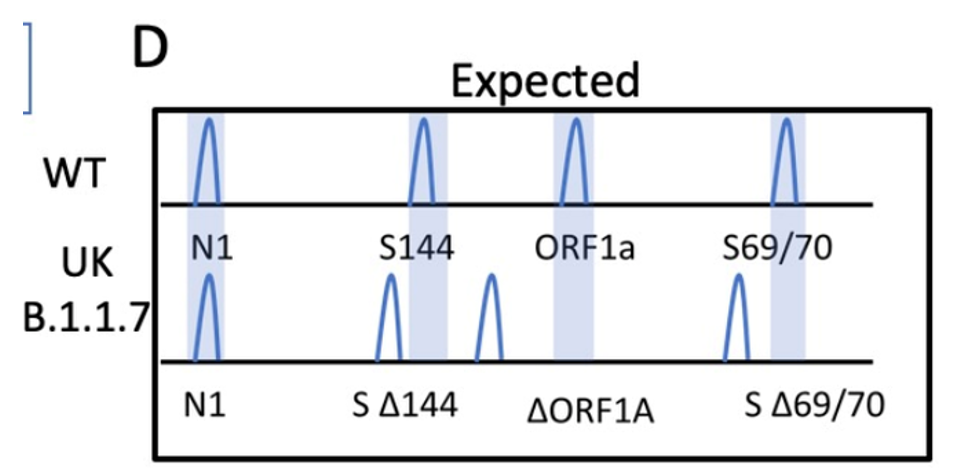
Theoretical picture of what the fragment analysis assay would look like for B.1.1.7. An actual patient sample results below, which showed the expected deletions exactly as predicted:
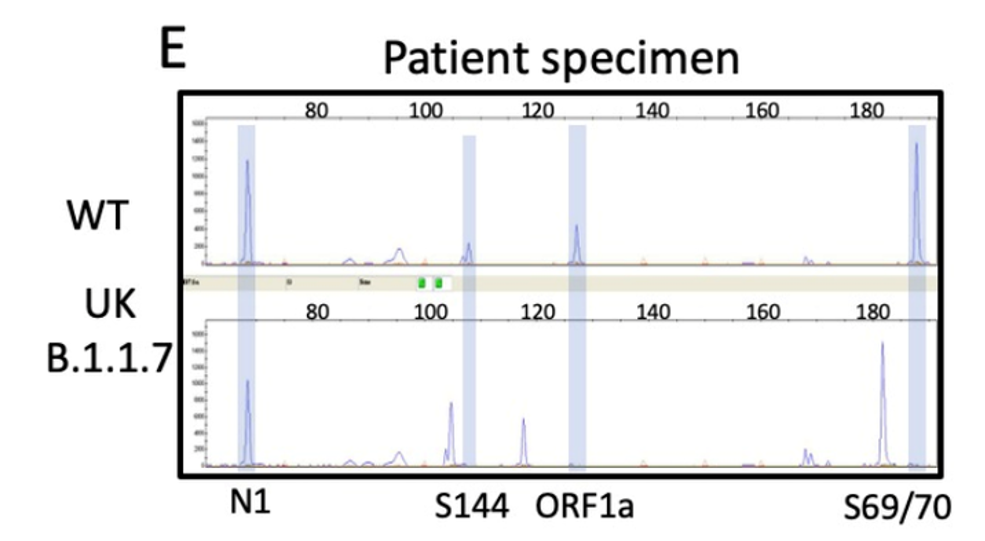
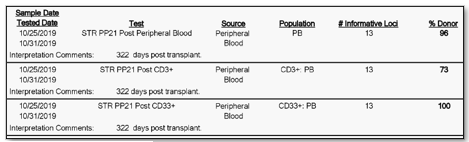
We have tested and sequenced over 500 positive specimens, and we found increasing levels of the B.1.1.7 strain prevalence up to nearly 30% by the middle of March. All screened B.1.1.7 specimens were validated by WGS. These results and the ability to detect the New York and California variants are detailed in our recent pre-print.
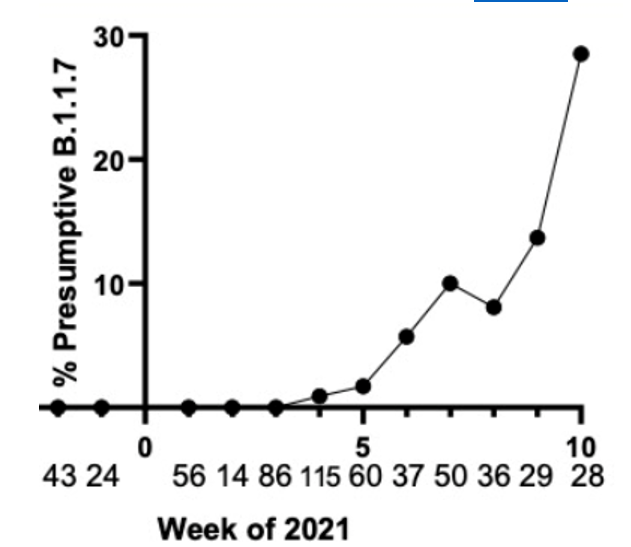
Weekly prevalence of isolates consistent with B.1.1.7 in North Texas.
Implications for future Variant Surveillance
As B.1.1.7 has become the dominant strain, and sequencing efforts are increasing. I would argue that assays should be used for what they are best at. For instance, it could be considered a waste of NGS time and resources to sequence all Variants when >50% are going to be B.1.1.7 if other tests can verify the strain faster for 10-20% of the cost. Instead, I think WGS should be focused on discovering emerging variants for which it is best suited. Across the US, case numbers have been decreasing and the number of specimens testable could be expanded by using a more sensitive PCR assay that could.
References
- Clark AE et al. Multiplex Fragment Analysis Identifies SARS-CoV-2 Variants. https://www.medrxiv.org/content/10.1101/2021.04.15.21253747v1
- Zhao Y et al. A Novel Diagnostic Test to Screen SARS-CoV-2 Variants Containing E484K and N501Y Mutations. A Novel Diagnostic Test to Screen SARS-CoV-2 Variants Containing E484K and N501Y Mutations | medRxiv
- Banada P et al. A Simple RT-PCR Melting temperature Assay to Rapidly Screen for Widely Circulating SARS-CoV-2 Variants. A Simple RT-PCR Melting temperature Assay to Rapidly Screen for Widely Circulating SARS-CoV-2 Variants | medRxiv
- Annavajhala MK et al. A Novel SARS-CoV-2 Variant of Concern, B.1.526, Identified in New York. A Novel SARS-CoV-2 Variant of Concern, B.1.526, Identified in New York | medRxiv
- Matic N et al. Rapid detection of SARS-CoV-2 variants of concern identifying a cluster of B.1.1.28/P.1 variant in British Columbia, Canada. Rapid detection of SARS-CoV-2 variants of concern identifying a cluster of B.1.1.28/P.1 variant in British Columbia, Canada | medRxiv
- Vogels CBF et al. PCR assay to enhance global surveillance for SARS-CoV-2 variants of concern. PCR assay to enhance global surveillance for SARS-CoV-2 variants of concern | medRxiv

–Jeff SoRelle, MD is Assistant Instructor of Pathology at the University of Texas Southwestern Medical Center in Dallas, TX working in the Next Generation Sequencing lab. His clinical research interests include understanding how lab medicine impacts transgender healthcare and improving genetic variant interpretation. Follow him on Twitter @Jeff_SoRelle.