Some very exciting news was recently announced about Artificial Intelligence impacting protein structure prediction. But like many of us, you probably thought, “Oh that’s nice.” Followed by either, “But that’s unlikely to impact lab medicine” or “I have no idea how they did that.” Today I will help turn around those last two thoughts for you!
The big news was that a U.K. company specializing in artificial intelligence, DeepMind (owned by Google-of course), won the CASP14 competition. CASP14 is the 14th edition of the biannual bake-off competition where teams use bioinformatic approaches to predict protein structures. The organizers then judge how well predictions match experimentally derived structures using a score called: GDT. This score reflects the distance of where something is vs. where it should be. Each of the ~150 protein sequences are scored on this basis and given a final percent identity score (0-100%).
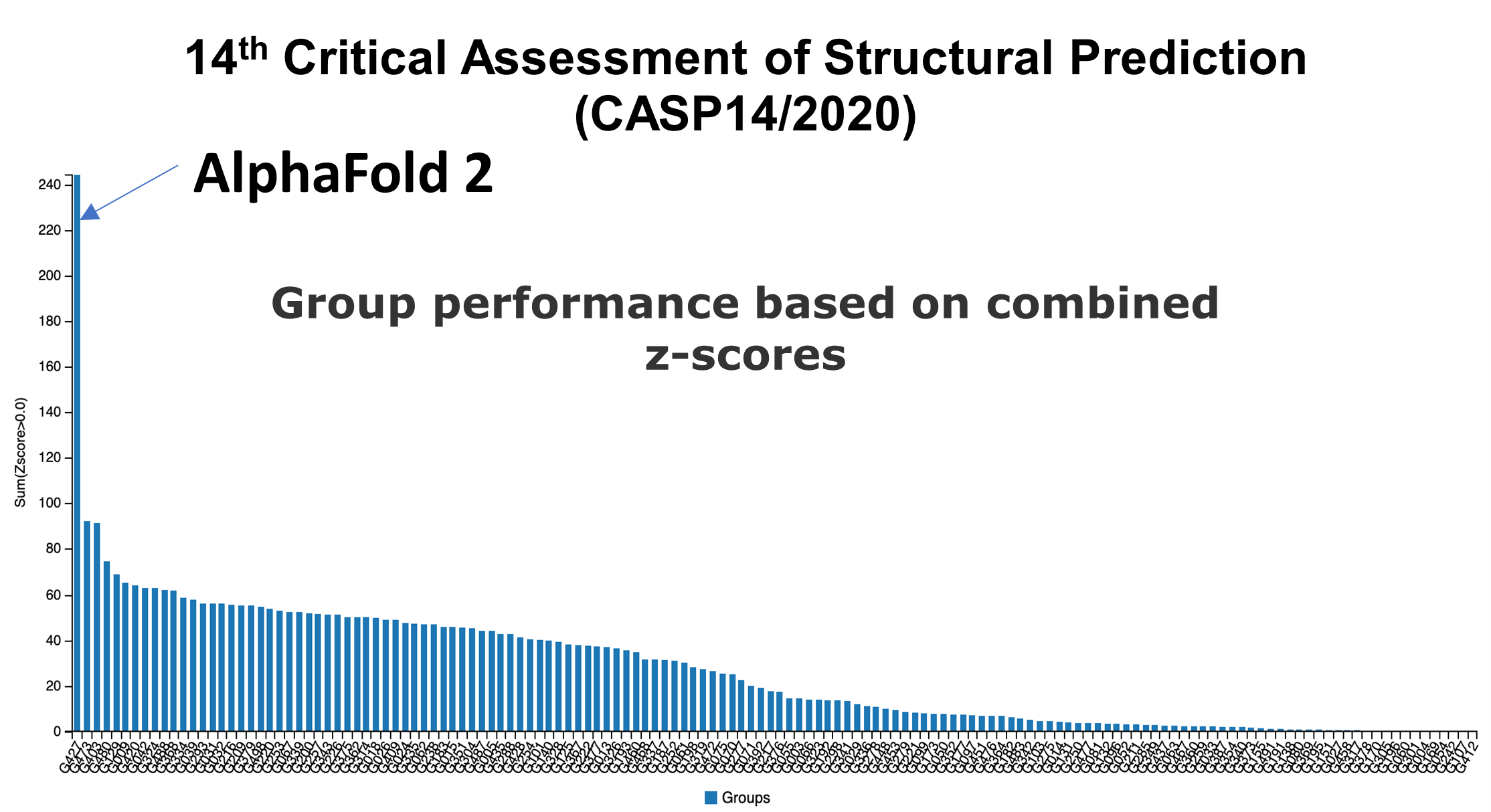
Since the competition started in 1997, the winners have scored ~50% on average. That is until 2 years ago when AlphaFold, the AI created by DeepMind, won with a top score of 55%. Their paper was published open access (Ref: https://www.nature.com/articles/s41586-019-1923-7) and used similar techniques applied by others where proteins were progressively folded by a computer until the lowest energy state is revealed.
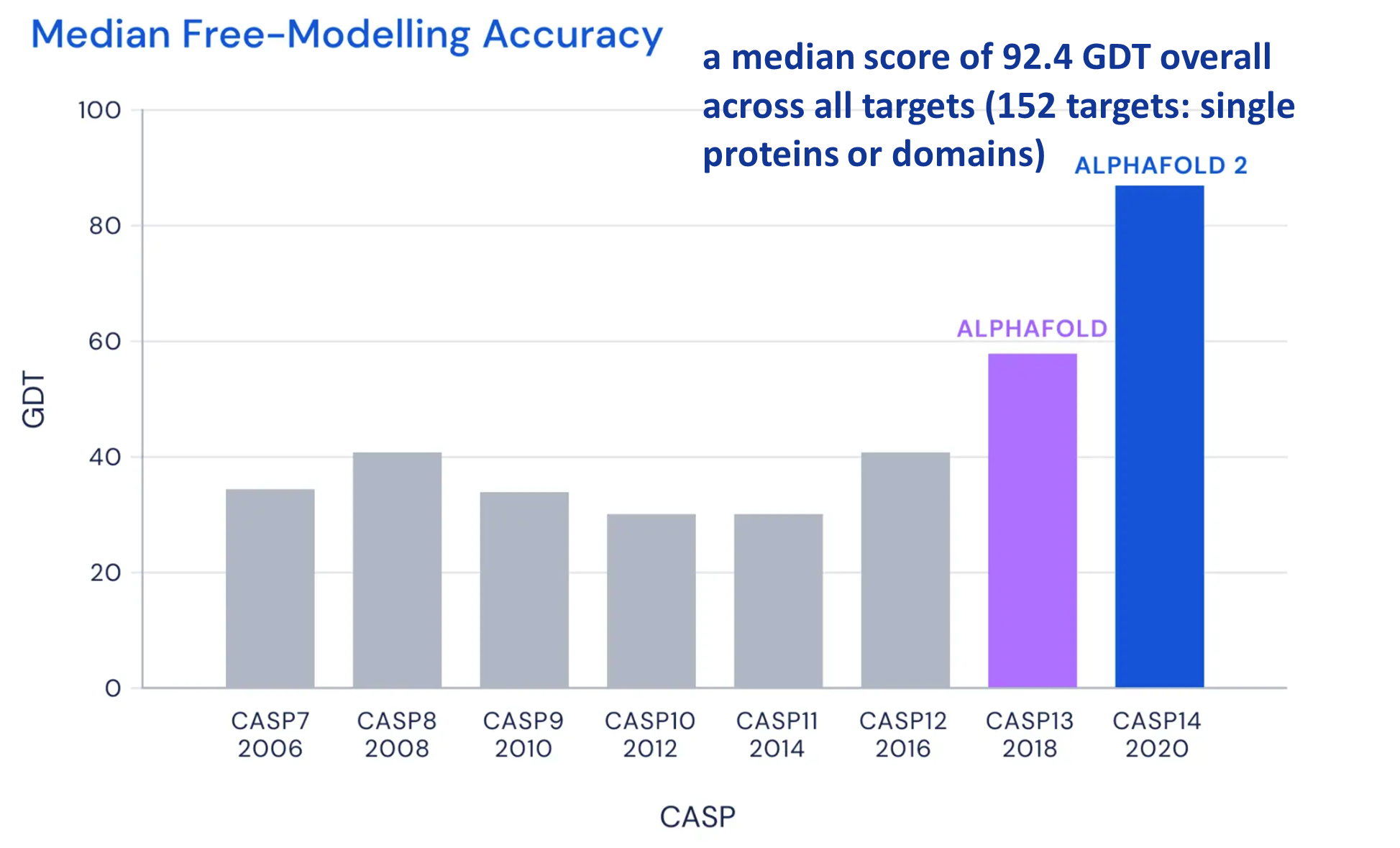
GDT: Global Distance Test (0-100); the percentage of amino acid residues within a threshold distance from the correct position. GDT of around 90 is considered competitive with results obtained from experimental methods.
The programs driving this folding may consider amino acid charge, size, and polarity, genetic conservation (Ref), or similarity to other protein domains. However, the innovation here was that DeepMind used artificial intelligence to examine sequence information with a convolutional neural network to identify structural constraints that are used to predict accurate protein folding.
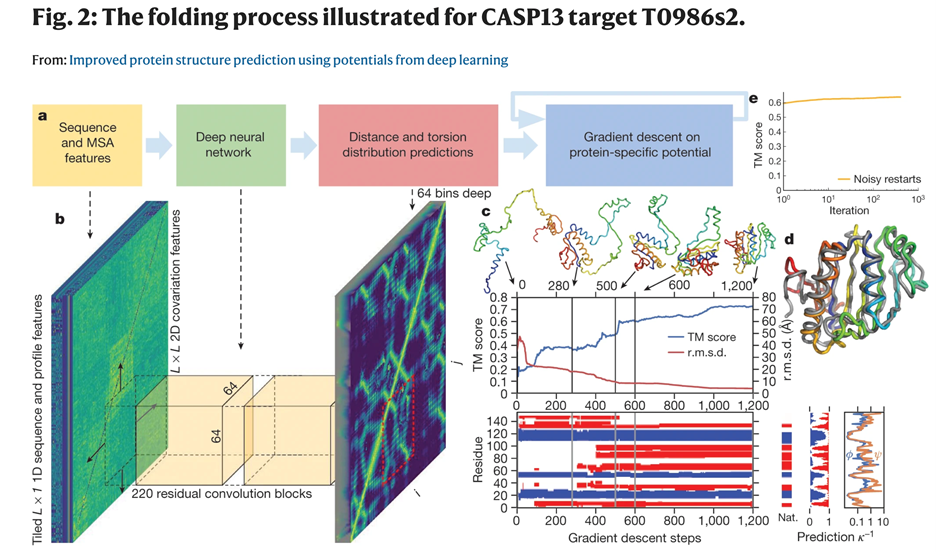
Figure 3. Sequence of events from Dataà Deep neural network (Artificial intelligence)àPredictions à protein folding process. (Figure 2 of this reference: https://www.nature.com/articles/s41586-019-1923-7/figures/2).
This results in one of those famous algorithms you’ve heard about. However, these algorithms are more complex than a simple linear regression and it is nearly impossible to trace how exactly how different levels of importance were assigned to each variable. An important requirement for an accurate A.I. derived algorithm is that it has a large training data set. Fortunately for Deepmind, they were able to train AlphaFold using about 170,000 structures that were determined experimentally using x-ray crystallography, nuclear magnetic resonance spectroscopy, and electron microscopy.
Although we haven’t seen what was changed between AlphaFold and AlphaFold 2, we have learned that AlphaFold 2 vastly outperformed the original in CASP14 with 91% accuracy. When programs are >90% accurate they are considered to be essentially as good as experimentally derived structures. In fact, AlphaFold 2 was able to provide more information than the experiments! One researcher found that their experimentally derived structure had a different configuration than the one predicted by AlphaFold, so they assumed the prediction by AlphaFold 2 was incorrect. After further analysis, the experimentally derived structure was found to be very similar to the structure predicted by AlphaFold 2. In another case, AlphaFold 2 predicted that an amino acid was in an infrequently found conformation, so they figured AlphaFold 2 made a mistake. After reanalyzing the experimental data, they found that that AlphaFold 2 was correct. It was even suspected that several lower-scoring structures based on NMR data may reflect lower accuracy in the experimental structure instead of a problem with the algorithm.
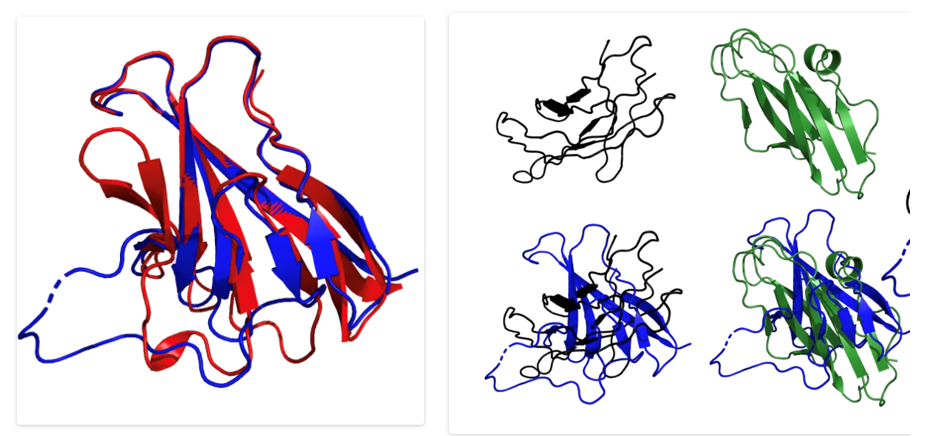
Will AI replace experimental crystallography? To answer this question, I turned to a colleague in my basic science lab, Lijing Su, who has been a structural biologist for many years. Like many cases of AI, this is a useful tool, but it doesn’t entirely replace her work because a lot of the structural biology research focuses on how proteins move and change as they do their job. Structural biology has moved beyond structures of single proteins and is now focused on how different proteins interact. There is still a role for crystallographers as AlphaFold cannot perform this role…yet.
All this still begs the question of a laboratorian “Who needs to know protein structure anyways?” We understand that knowing protein structures can help explain function, which has implications with drug development. However, our main role is to provide tests that diagnose disease. A major challenge in molecular pathology is to predict whether a genetic variant causes loss of protein function. Current software has poor performance (PolyPhen2 sensitivity= 45% specificity= 50%) as they mainly measure changes in chemical properties and amino acid site conservation. One potential application of AlphaFold is to examine the effect of genetic variants on protein structure. Pathogenic changes would be predicted to deform portions of the structure impairing activity or provoking degradation through the unfolded protein response.
As the current speed of the program is quite long, this could be difficult to implement immediately, but it is imaginable that this will become quicker. A straightforward way to validate this AI software would use confirmed pathogenic or benign variants from the public database ClinVar. There are over 1,000,000 entries into this database, which would provide a useful training and validation set. It is likely that change in protein structure would be a stronger mechanism of disease for certain types of proteins (ion channels for epilepsy or myosin chains for muscular disorders) and a less strong predictor of pathogenicity for other types of proteins (enzymes for metabolic disorders or signaling proteins where protein-protein interaction is important for function).
This blog entry was written with the very helpful insights and knowledge of Lijing Su, PhD.
References
- Senior AW et al. Improved protein structure prediction using potentials from deep learning. Nature. 2020; 577: 706–710.
- CASP14 website: https://predictioncenter.org/casp14/
- Arnold CN et al. ENU-induced phenovariance in mice: inferences from 587 mutations. BMC Res Notes. 2012; 5: 577.
- https://deepmind.com/blog/article/alphafold-a-solution-to-a-50-year-old-grand-challenge-in-biology
-Lijing Su is Assistant Professor in the Center for Genetics of Host Defense at the University of Southwestern Medical Center. She specializes in structural biology and helps determine multi-protein interactions to explain unknown mechanisms of genes important to immunology.

–Jeff SoRelle, MD is Assistant Instructor of Pathology at the University of Texas Southwestern Medical Center in Dallas, TX working in the Next Generation Sequencing lab. His clinical research interests include understanding how lab medicine impacts transgender healthcare and improving genetic variant interpretation. Follow him on Twitter @Jeff_SoRelle.
