A 72 year old man was admitted to the hospital for an aneurysm repair. The physician ordered a type and crossmatch for 6 units of blood in preparation for surgery. The patient history included surgery in 2016 during which he was transfused with 4 units of RBCs.
patient’s blood type: A positive
antibody screen: negative
history: anti Jkb (2016)
6 Jkb negative units were found and full crossmatches were performed. One of the 6 donor units was incompatible. What is the most probable explanation for these findings?
If the patient has a negative antibody screen, and no history of an antibody, most facilities would do an electronic crossmatch or an immediate spin crossmatch. The immediate spin (abbreviated) crossmatch will simply verify ABO compatibility. However, if the patient has a positive antibody screen, we must identify the antibody, phenotype the patient, and do a full AHG crossmatch with donor units that are antigen negative for the corresponding antibody. In this case, the patient had a history of an antibody, so the antibody must be honored, and antigen negative units must be chosen for transfusion.
Kidd antibodies demonstrate dosage, are often weak, and can be found in combination with other antibodies. Because if this, they can be notoriously difficult to detect. They are usually IgG and are made in response to transfusion or pregnancy. Jkb has an antigen frequency of about 73% in the white population and about 43% in the black population. To find antigen negative blood, we consider that about 27% of units would be antigen negative. The tech working on the sample screened 21 units and found 6 that were Jkb negative.
AHG crossmatch results:
unit 1: compatible
unit 2: compatible
unit 3: compatible
unit 4: 3+ at AHG
unit 5: compatible
unit 6: compatible
There are 2 possible scenarios for the above results. A crossmatch is a test between donor’s red blood cells and patient’s plasma. Antigens, we know, are on red blood cells and antibodies are detected in the plasma. So, with a negative antibody screen, crossmatch incompatibility is due either to a patient antibody to a low incidence antigen on the donor red blood cells, or a donor cells with a positive direct antiglobulin test. We can easily rule in or out a positive donor DAT by performing a DAT on the segment. If the donor unit has a positive DAT, the unit should be quarantined and the positive DAT reported to the collecting facility. If the donor unit has a negative DAT, the patient likely has an antibody to a low incidence antigen.
Low frequency antigens are uncommon, but antibodies that recognize them are less rare. Fortunately, for patients with these antibodies to low frequency antigens, finding antigen negative compatible blood is easy. As we can see, 5 of the 6 chosen units were negative for the unknown low frequency antigen and were antiglobulin crossmatch compatible. The low prevalence of the antigen makes compatible blood readily available. If transfusion is necessary, it should not be delayed while waiting for identification of the antibody.
In this case, the antibody screen was repeated and the negative result was verified. In many cases, it may not be possible for a lab to identify the antibody because the lab may not have the necessary panel cells or typing reagents. Yet, these antibodies to low incidence antigens that react at AHG can be clinically significant and cause severe hemolytic transfusion reactions. To identify the antibody, you may need to send the sample to a reference lab for testing against a panel of reagent red cells that express low incidence antigens. Alternately, the donor red cells that were incompatible can be tested against known antibodies to low prevalence antigens to help identify the antibody.
In this patient, anti-Wra was identified. The incompatible donor unit was verified to be Wra positive. Wra is part of the Diego system, usually IgG, and has ben implicated in hemolytic transfusion reactions.
One of the reasons I have written up this case is questions my Transfusion Medicine students often ask about exam and exam prep questions concerning incompatibility. Below are 2 questions to give examples of the confusion.
“At the indirect antiglobulin phase of testing, there is no agglutination between patient serum and screening cells. One of 3 donor units was incompatible.. The most probable explanation for these findings is that the:
patient has an antibody directed against a high incidence antigen
patient has an antibody directed against a low incidence antigen
donor has an antibody directed against donor cells
donor has a positive antibody screen”5
answer: b
“Which of the following would most likely be responsible for an incompatible antiglobulin crossmatch?
recipient’s red cells possess a low incidence antigen
anti-K antibody in donor serum
recipient’s red cells are polyagglutinable
donor red cells have a positive direct antiglobulin test”4
answer: d
I am asked why is one answer “low prevalence antigen” and one answer “positive DAT”? I typically ask questions of my students to let them reason out the answer. Take a careful look at the words antigen and antibody. Remember that a DAT is a test of red cells, the IAT tests for antibodies in plasma. A crossmatch uses donor red cells against patient plasma. Therefore, even though these are both reasons for the incompatibility of one out of multiple units, each question only has one answer of a common reason for such incompatibility. Be sure to read questions and use your theory and knowledge of testing when encountering discrepancies and problems in Blood Bank. To all of my students: Happy Studying for your ASCP exam!
References
Fung, Mark K., Technical Manual 18th ed, Bethesda: AABB, 2014.
Harmening, Denise M. Modern Blood Banking and Transfusion Practices, 7th edition, 2019.
Schonewille, Henk, et al. “The importance of antibodies against low‐incidence RBC antigens in complete and abbreviated cross‐matching”. The Journal of AABB. 20 June 2003.
-Becky Socha, MS, MLS(ASCP)CM BB CM graduated from Merrimack College in N. Andover, Massachusetts with a BS in Medical Technology and completed her MS in Clinical Laboratory Sciences at the University of Massachusetts, Lowell. She has worked as a Medical Technologist for over 30 years. She’s worked in all areas of the clinical laboratory, but has a special interest in Hematology and Blood Banking. When she’s not busy being a mad scientist, she can be found outside riding her bicycle.
Good morning!
We’re entering the holiday season, and it’s an exciting time for all. I love
seeing the ethnic and cultural diversity as we all celebrate our favorite
holidays with family and friends. I myself look forward to the holiday season.
It’s a festive time and a season of giving and sharing. It’s a favorite time of
year to share traditions and create new ones. However, at a time when stores
have Christmas candy on the shelves, holiday lights up and holiday music playing
on the day after Halloween, I feel a bit rushed and want to slow down and find
better ways to celebrate and enjoy the season. Over the past few years I have
been making a special effort to become more environmentally conscious;
remembering my reusable bags at stores, purchasing more reusable products, and
reusing, recycling, and upcycling whenever I can. I belong to a community ‘buy
nothing’ group and am warmed by the generosity of strangers to others in the
community. It’s wonderful to give from our abundance and to receive wish list
items from neighbors without having to exchange money. And it’s great for the
environment, too. Used items are being put to use by others, and not into
landfills. People in the community have asked for or gifted furniture,
clothing, tools, toys and many other goods and services. I have gifted no
longer needed clothing, household items, excess fabric from my fabric stash,
and donated my time to participate in a career fair at a local high school. I
have been given a car set for my grandchildren when they visit, toys, and
someone even loaned me a bike trailer so we could take my granddaughter out for
a bike ride. The generosity makes it feel like the holiday season all year
round.
So, you may be
asking, “where is this blog going?” I saw a memo from Red Cross this week that
there is a critical need for blood and platelets and thought that giving to our
community with the gift of blood would be a wonderful way to make this holiday
season even better! It’s one of the most generous gifts we can give, and costs
nothing. Every 2 seconds in the US, someone needs a blood product. That’s about
36,000 units of red blood cells, 7,000 units of platelets and 10,000 units of
plasma needed every day. 21 million blood products are transfused every year.1
That’s a lot of blood. And, these blood
products cannot be manufactured, so must come from volunteer donors.
In
the US, we need to collect about 13,000 units a day to meet demand. Approximately
14 million units of whole blood are collected each year from roughly
7 million donors.1 The blood is processed into components and
used in the treatment of surgical, obstetric, oncology, and other patients. One
unit of whole blood can be made into up to 3 components and used to help up to
3 patients. Yet, even with all these donations we still cannot keep up with
demand. Weather, holidays, illness and travel can all affect blood donations. Shortages
are not just apparent during the winter holiday season. This past summer, the
Red Cross announced a critical blood shortage around the July 4th
holiday. Compared to other weeks, there were 17,000 fewer blood donations
during the week of July 4th. As of July 9, the Red Cross had less than a
three-day supply of most blood types and less than a two-day supply of Type O
blood. 2 During the summer, and particularly during the holiday
week, people are busy with other activities or traveling. In the winter, busy
schedules, holiday travel, winter weather and seasonal illnesses contribute to
fewer blood and platelet donations. Severe weather can also cause the
cancellation of blood drives which greatly impact the blood supply.
Some people donate blood because they see this critical need
and hear the calls for blood. Others donate because a classmate or friend asked
them to. Some people feel it’s their civic duty. For some, it just makes them
feel good to help another person. And, others donate for the cookies and tee
shirt. Yet, for all donors, it is a form of volunteerism and giving to the
community. But, did you know that, other than the benefits from helping others,
there are benefits to the donor, as well? Helping others can improve our
emotional and physical health. It can help reduce stress, improve emotional well-being
and help people feel a sense of belonging. A study conducted in Sweden
concluded that regular blood donors enjoy better than average health.Blood
donors had an overall mortality 30% lower and a cancer incidence 4% lower than
the control population.3 Donating blood may help reduce high iron
stores, a risk factor for heart attack. In addition, there have been several
studies over the past few years, exploring the hypothesis that regular blood
donations may help in the management of hypertension and high cholesterol.
Another
interesting benefit of blood donation is being able to contribute to science
and research. For example, there is currently a study being conducted on donor
blood to test an investigational nucleic acid test for Babesia microti. Babesia
microti is responsible for most transfusion-transmitted babesiosis cases in the
United States, but there is no licensed test for screening for B. microti in
donated blood. Participation in this study can help obtain FDA approval for a
screening test. By giving
your consent to use your blood sample, there is no additional blood taken and
no further time commitment, but you can help protect the public health by
supporting the development of a new blood safety test.
How
can we, as individuals, help? About 38% of the population is eligible to donate
blood, but less than 10% of the population actually donates. To be eligible to
donate, you should be in good general health and feeling well. You
must be at least 17 years old in most states
(16 years old with parental
consent in some states) but there is no age limit to donation. Adult doors must
weigh 110 lbs, but there are additional height and weight requirements for
donors 18 years old and
younger. There have also been some recent changes to blood donor requirements. I
will not be able list all of them here, but some of them don’t change a
deferral, only the reasoning behind the deferral. One of the most prominent
changes is, as of 2016, the indefinite deferral for men who have had sex with
men, has been changed to a 12 month deferral since the last sexual contact with
another man . Also changed is the minimum hemoglobin for male donors. This has been
raised from 12.5g/dl to 13.0 g/dl. Until this time, the cutoff was the same for
both males and females. Males with a Hgb below 13.0 g/dl are considered anemic
and are no longer eligible to donate blood. On the other hand, the criteria for
females to be mildly anemic is a Hgb below 12.0 g/dl, so females between 12.0
g/dl and 12.5 g/dl, though not considered anemic, are still not eligible to
donate. The minimum hemoglobin for females has not changed and remains 12.5
g/dl. To review other eligibility requirements, visit https://www.redcrossblood.org/donate-blood/how-to-donate/common-concerns/first-time-donors.html
So, in this busy season, we often find ourselves with little time to get our own “to do” lists done, yet alone volunteer our time for others. But most of us would welcome an hour to reduce stress and improve our emotional well-being. Please consider a gift of self this season. It takes about an hour of your time, you get to sit and relax with your feet up, to feel good about yourself, and you’ll even get a snack!
Edgre, G et al. Improving health profile of blood donors as a
consequence of transfusion safety efforts. Transfusion. 2007 Nov;47(11):2017-24.
Kamhieh-Milz
S, et al.Regular blood donation may help in the
management of hypertension: an observational study on 292 blood donors. Transfusion. 2016 Mar;56(3):637-44. doi: 10.1111/trf.13428.
Epub 2015 Dec 8.
-Becky Socha, MS, MLS(ASCP)CM BB CM graduated
from Merrimack College in N. Andover, Massachusetts with a BS in
Medical Technology and completed her MS in Clinical Laboratory Sciences
at the University of Massachusetts, Lowell. She has worked as a Medical
Technologist for over 30 years. She’s worked in all areas of the
clinical laboratory, but has a special interest in Hematology and Blood
Banking. When she’s not busy being a mad scientist, she can be found
outside riding her bicycle.
In my July post, “Blood Bank Case Study:
What’s Your Type?” I discussed some of the dilemmas when dealing with a weak D
phenotype and the fact that there is no standard or general consensus as to the testing
performed or terminology to be used in resulting a weak D patient. Results
obtained on patient testing also vary depending on the method used, and the
anti-D reagent and enhancement used in testing. This can be confusing to medical
technologists, physicians and to patients.
For anyone who has not been in the Blood bank for a while, the Du variant was first
recognized in 1946 and renamed weak D in 1992. To
review last month’s blog, serologic studies have distinguished three broad
categories of D variants, weak D, partial D, and DEL, from conventional D. A
serologic weak D phenotype is one that has no or weak reactivity (≤2+) of RBCs
with an anti‐D reagent at immediate spin, but does agglutinate with antihuman
globulin. Since there is no general consensus on how labs perform
and report patient testing for weak D, it is left up to individual
interpretation as to what type blood these patients should receive, and, if
pregnant, if they should receive Rh D immune globulin (Rhogam). Last month I
focused on testing, resulting, donors and blood administration. In this blog I
will focus on issues concerning weak D in the obstetric population and how labs
can move forward now and in the future towards the best patient care and blood
management.
About 15% of
Caucasians are RhD negative. About 3% are weak D phenotypes.In the genral
population, this means that about 0.2% to
1.0% inherit RHD genes that code for serologic weak
D phenotypes.2 In Europe
and the US, weak D phenotypes are the most common D variants found, but we also know that the
prevalence of weak D phenotypes varies by race and ethnicity. Today we have
much more information about D antigen expression than we had in the past, because
we have the availability to genotype these weak D RBCs. We know that more than
84 weak D types have been identified, but types 1, 2, and 3 account for more
than 90% of these in people of European ethnicity.1 Currently,
with the mixed ethnicity population in the US, about 80% of people who inherit RHD genes for serologic weak D
phenotypes are found to be weak D
type 1, 2, or 3.3 We also know that
types 1, 2 and 3 are unlikely
to become alloimmunized to anti-D, so they can safely be treated as RhD
positive and receive RhD positive units.
The introduction of RhD immune globulin in 1968is one of the great success
stories in obstetrics. Rhogam has been used very successfully in developed
countries in the prevention and treatment of hemolytic disease of the fetus and
newborn due to RhD alloimmunization. The routine recommendation is that women
who are candidates for Rhogam receive one dose at approximately 28 weeks’
gestation and a second dose after the delivery of an Rh pos baby. Additional recommendations are for administration
of Rhogam after threatened miscarriage, abdominal trauma during pregnancy and
before invasive diagnostic procedures.
But,
who is a candidate? Any unsensitized woman who is RhD negative and who may be
carrying or who delivers an RhD positive baby is a candidate for Rhogam. And,
that brings us back to the problem that we have no standardization for the
reporting of serological weak D phenotypes.
As
an example, let’s look at a patient who has 3 children. Many labs do not do
weak D testing on patients and report anyone who is RhD negative at immediate
spin as RhD negative. This patient was typed at such a lab (Lab #1) as RhD
negative, and received Rhogam for her first pregnancy. During her 2nd
pregnancy, she had moved to a different state, and went to another lab (Lab #2)
for prenatal testing. This lab performed serologic weak D testing and found
this patient to be weak D positive and reported her type as RhD positive.
Rhogam was not further discussed during this pregnancy and the patient did not
receive Rhogam. The patient had blood drawn during her 3rd pregnancy
at yet a third hospital (Lab #3). Some labs distinguish women who are pregnant
or of childbearing age from the general population, and have different procedures
on the reporting of RhD type on these women. This hospital’s procedure was to
do weak D testing on all patients, but, in women of childbearing age, if weak D
positive, they report these women as RhD negative. The patient was told she was
RhD negative and would be a candidate for Rhogam. At this time the woman
thought she remembered that she didn’t get Rhogam with her second pregnancy and
was a little confused, but with 2 young children and pregnant with her 3rd,
she simply followed the doctor’s recommendation and didn’t question further.
When her 3rd child was 4 months old, she attended a Red Cross blood
drive at work and donated a unit of blood. Soon she received a blood donor card
in the mail that said she was RhD positive. At this point she was thoroughly
confused and questioned all the lab results she had had done over the past 6
years. On her next visit to the doctor she questioned her obstetrician. The
obstetrician recommended RhD genotyping. The woman was found to be weak D type
2. The doctor explained to her that all blood donors who are weak D are treated
as RhD positive, but, that as a patient, policies and procedures vary. However,
he also informed her that now that they had her genotype, she would be
considered RhD positive. He explained that the genotype was DNA testing, would
not need to be repeated, she would not need Rhogam for any future pregnancies
and she could safely receive RhD positive blood products.
The
American College of Obstetricians and Gynecologists (ACOG) guidance practice
bulletin of 1981 recommended that recommended that RhD‐negative women “whether Du positive or
Du negative” were candidates for Rhogam. Shortly
afterwards, that recommendation was
reversed and revised to read “[a]
woman who is genetically Du‐positive is Rh‐positive and
administration of Rh immune globulin is unnecessary.1 This remained the recommendation of the
group until the latest version of this publication in 2017. The 2017 ACOG
guidelines recommend giving Rhogam to weak D positive patients, “in appropriate
clinical situations, until further studies are available.”3 Another comparative
study published in 2018 reported inconsistency between national groups over how
to treat weak D phenotypes and recommended the creation of international
guidelines.4
Thus,
the controversy over whether a pregnant woman who is weak D positive is RHD
positive or RHD negative continues. The latest recommendations, and those of
ACOG, are for a move to genotyping patients with a serological weak D
phenotype. There are several benefits to this. As we can see from my case study
example, genotyping put this woman at ease and gave her definitive answers
about her blood type. It also can do the same thing for medical technologists
and physicians. RHD genotyping only needs to be performed once on a
patient. If performed at the first prenatal appointment, this would alleviate
much confusion as to procedures and how to report the results. I have in blood
bank, that whenever we have a weak D on a prenatal patient, there are questions
about how to result them, and we refer to the SOPs. We also occasionally get a
patient who had previously been typed elsewhere where the reporting procedures
were different and there is therefore an apparent discrepancy between the
current and historical typing. This causes frequent phone calls from physicians
and nurses asking for clarifications on weak D types, and questions about
Rhogam. Lastly, RHD genotyping could avoid confusion which could lead to
transcription and computer entry errors when entering types on these patients. RHD
genotyping would solve all of these problems and eliminate confusion.
Additional
benefits of RHD genotyping are, if RHD genotyping was performed
on all weak D transfusion recipients, we could save as many as 47,700 units of
RHD negative RBCs annually.3 With the availabilityof
molecular testing, there is no reason to administer RhD negative units to
patients who can use RhD positive units. This could help alleviate the constant
shortage of RHD negative units. With RHD molecular testing, these
critical units could be reserved for patients who are truly RhD negative.
It may not be feasible for all laboratories to perform molecular testing for RHD genotypes, but reference laboratories should offer affordable testing for the most prevalent and clinically relevant RHD genotypes. From a study done of over 3100 laboratories, it was found that, at this time in the US, most labs are managing weak D phenotypes as RhD negative. Laboratories not performing weak D testing are essentially avoiding their detection. Clinical laboratories should instead increase the detection of serological weak D and interpret these with the use of RHD genotyping. Rhogam shortages exists, and RHD genotyping could save thousands of injections of Rhogam annually in the US alone, and at the same time, avoid the unnecessary administration of products to patients. The work group study calculated that annually, approximately 24,700 doses of unnecessary Rhogam could be avoided.1 It is time to move forwards to molecular testing for the best patient care and blood management.
References
Sandler SG, Flegel, WA, Westhoff CM, et al. It’s time to phase in RHD genotyping for patients with a serologic weak D phenotype. Transfusion 2015;55:680‐9
Garratty G. Do we need to be more concerned about weak D antigens? Transfusion 2005;45:1547‐1551.
Practice Bulletin No. 181: Prevention of Rh D AlloimmunizationObstetrics & Gynecology: August 2017 – Volume 130 – Issue 2 – p e57-e70 doi: 10.1097/AOG.0000000000002232
Sperling, JD et al. Prevention of RhD Alloimmunization: A Comparison of Four National Guidelines. Am J Perinatol. 35(2):110-119. doi: 10.1055/s-0037-1606609. Epub 2017 Sep 14.
-Becky Socha, MS, MLS(ASCP)CM BB CM graduated
from Merrimack College in N. Andover, Massachusetts with a BS in
Medical Technology and completed her MS in Clinical Laboratory Sciences
at the University of Massachusetts, Lowell. She has worked as a Medical
Technologist for over 30 years. She’s worked in all areas of the
clinical laboratory, but has a special interest in Hematology and Blood
Banking. When she’s not busy being a mad scientist, she can be found
outside riding her bicycle.
The general public doesn’t always know a lot about laboratory
testing in general, but most people know a little about blood types, even if
it’s what they have learned from TV! Blood types do seem to come up in casual
conversation. We might hear a conversation about blood type after someone has
donated blood, or between family members comparing notes, who ask “What’s your
type?” Yet, even with medical technologists, there can still be some confusion
about blood types and blood typing, particularly if one has not worked in Blood
Bank in many years. I recently received an email from a colleague who had a few
questions about blood types, as she has not worked in Blood Bank for over 40
years. I always tell my students that no question is a bad question, and indeed,
she asked some very good questions, which I will address with this case study.
What blood type is listed on a patient’s chart if they type “O
Du”?
What blood type is recorded on a donated unit of blood typed “O
Du”?
What type of blood does an “O Du” patient receive?
Can an “O Du” patient have a transfusion reaction if they are
transfused with O positive blood? Would she need to receive O negative blood in
a transfusion?
Does an “O Du” patient need to receive RhoGAM if she pregnant and
her husband is Rh positive?
If you have ever wondered or can’t remember details about any of
these questions, you’re in the right place. So, what’s new, if anything, with
blood types?
Landsteiner discovered the ABO blood group system in 1901, and
identified A, B and O blood types, using experiments performed on blood from
coworkers in his laboratory. The discovery of the codominant AB blood type soon
followed, but it was not until around 1940 that the Rh blood group was first described.
In 1946, Coombs and coworkers described the use of the antihuman globulin (AHG)
to identify weak forms of Rh antibodies in serum. For us old blood bankers, the
original name for this test was the Coombs’ test. (You will still find
physicians ordering a Coombs’ test!) The current and proper name for this is
the direct antibody test (DAT), which is used to detect in vivo sensitization
of RBCs. AHG can also be used to detect in- vitro sensitization of RBCs using
the 2 stage indirect antibody test (IAT).
Since Landsteiner’s work, we have not discovered any new blood groups
that are part of the routine blood type. The ABO and Rh blood groups are still
the most significant in transfusion medicine, and are the only groups consistently
reported. However, we currently recognize 346 RBC antigens in 36 systems.1 Serological tests determine RBC
phenotypes. Yet, today we can also determine genotype with family studies or
molecular testing. This case study and 2 part blog reviews some terminology in
phenotyping, some difficulties and differences encountered, and explores the
possibility of RHD genotyping to assess a patient’s true D status.
Our case study involves a 31 year old woman who is newly married.
She is not currently pregnant, has never been pregnant, is not scheduled for
surgery but has had a prior surgery 15 years ago, and has never received any
blood products. She and her husband recently donated blood and, as first time
blood donors, just got their American Red Cross (ARC) blood donor cards in the
mail. The husband noted that his card says that he is type O pos. The woman
opens her card, and, with a puzzled look on her face, says “My card says I’m an
O Pos, too. There must be a mistake.” She knows she has been typed before and
checks her MyChart online. Sure enough, her blood type performed at a local
hospital is listed in her online MyChart as O negative. She further checks
older printed records and discovers that 15 years ago, before surgery, she was
typed at a different hospital as “O Du”. She is very upset, wondering how she
can have 3 different blood types. She is additionally concerned because they
are planning to have children and recalls being told that because she is Rh
negative, that she would need Rhogam. Is she Rh negative or positive, and what
does Du mean? Will she need Rhogam when pregnant? She has many questions and calls
the ARC donor center for an explanation.
What blood type is listed on a patient’s chart
if they type “O Du”?
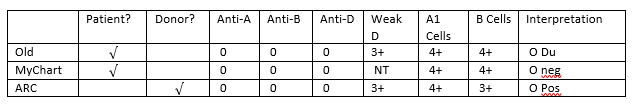
What is happening here, what is this woman’s actual blood type, and what testing can be done to ensure accuracy in Rh typing? From the patient reports, it appears that this woman has what today we call a “weak D.” Du is an older terminology that should no longer be used, and that has been replaced by the term “weak D.” But, why does she have records that show her to be an O neg, a type O, Du (today, this would be written O weak D), and now, a card from ARC stating she is O pos?
RhD negative phenotypes are ones that
lack detectable D antigen. The most common Rh negative phenotype results from
the complete deletion of the RHD gene. Serologic testing with anti-D is usually
expected to produce a strong 3+ to 4+ reaction. A patient with a negative
anti-D at IS and at IAT would be Rh negative. If the patient has less than 2+ strong
reaction at immediate spin (IS), but reacts at IAT, they would be said to have
a serologically weak D.1 Historically, weak D red blood cells (RBCs)
are defined as having decreased D antigen levels which require the IAT for
detection. Today’s reagents can detect many weak D
types that may have been missed in the past, without the need for IAT. However,
sometimes IAT is still necessary to detect a weak D. When this is necessary is
dependent on lab SOPs and whether this is donor testing or patient testing. The
reported blood type of this patient also depends on the SOPs of the laboratory
that does the testing. And, the terminology used for reporting is also lab
dependent. It is not required by AABB to test patient samples for weak D
(except for babies of a mother who is D negative). There is also no general
consensus as to the terminology to be used in reporting a weak D. Some labs would
result this patient as O negative, weak D pos. Some labs may result O pos, weak
D pos. Others may show the individual reactions but the resulted type would be
O pos. Labs who do not perform weak D testing would report this patient as O, Rh
negative. The following chart explains why this patient appears to have 3 types
on record.
Figure 1. Tube typing results of same patient from different labs with different SOPs.
What blood type is recorded on a donated unit
of blood typed “O Du?”
AABB Standards for Blood Banks and
Transfusion Services requires all donor blood to be tested using a method that
is designed to detect weak D. This can be met through IAT testing or another
method that detects weak D. If the test is positive, the unit must be labeled
Rh positive. This is an important step to prevent alloimmunization in a
recipient because weak D RBCs can cause the production of anti-D in the
recipient. This also explains why the ARC donor card this patient received
lists her type as O pos.
What type
of blood does an “O Du” patient receive?
Historically, weak D red blood cells
(RBCs) were defined as having decreased D antigen levels which require the IAT
for detection. A patient who is serologic weak D has the D antigen, just in
fewer numbers. This type of weak D expression primarily results from
single-point mutation in the RHD gene that encodes for a single amino acid
change. The amino acid change causes a reduced number of D antigen sites on the
RBCs. Today we know more about D antigen expression because we have the
availability to genotype these weak D RBCs. More than 84 weak D types have been
identified, but types 1, 2, and 3 represent more than 90% of all weak D types
in people of European ethnicity.2 An Rh negative patient has no D
antigen and should, under normal circumstances, only receive Rh negative blood.
Yet, there has been a long history of transfusing weak D patients with Rh
positive RBCs. 90% of weak D patients genotype as Type 1, 2 or 3 and may
receive Rh positive transfusions because they rarely make anti-D. 2
It is now known that weak D can actually
arise from several mechanisms including quantitative, as described above, position
effect, and partial D antigen. Molecular testing would be needed to
differentiate the types, but, with the position effect, the D antigen is
complete and therefore the patient may receive Rh positive blood with no
adverse effects. On the other hand, a partial D patient may type serologically
as Rh negative or Rh positive and can be classified with molecular testing. It
is important to note that these partial D patients are usually only discovered
because they are producing anti-D. If anti-D is found, the patient should
receive Rh negative blood for any future transfusions.
Thus, 3 scenarios can come from typing
the same patient. With a negative antibody screen, and because 90% of weak D
patients have been found to be Type 1, 2 or 3 when genotyped, many labs do not routinely
genotype patients and will report the blood type as Rh pos and transfuse Rh pos
products. However, depending on the lab medical director and the lab’s SOPs,
these same patients may be labeled Rh neg, weak D and receive Rh negative
products. There is no general consensus on the handling and testing of weak D
samples. The 3rd scenario is that many labs do not test for weak D
in patients at all, and a negative D typing at IS would result in reporting the
patient as Rh neg, with no further testing. In this case, the patient would be
transfused with Rh negative products.
Can an “O
Du” patient have a transfusion reaction if they are transfused with O positive
blood? Would she need to receive O negative blood in a transfusion?
This question was covered
somewhat in the above discussion. Policies regarding the selection of blood for
transfusion are lab dependent, dictated by the lab medical director, and are
based on the patient population, risk of developing anti-D, and the
availability or lack of availability of Rh negative blood products. Anti-D is
very immunogenic. Less than 1 ml of Rh pos blood transfused to an Rh negative
person can stimulate the production of anti-D. However, not all patients
transfused with Rh positive blood will make and anti-D. As discussed above, 90%
of weak D patients are types 1, 2 or 3, would be unlikely to become
alloimmunized to anti-D. If a weak D patient with a negative antibody screen
receives a unit of D pos RBCs, there is a very small possibility that they are
a genotype who could become alloimmunized to the D antigen and produce anti-D. However,
as stated above, the majority of weak D patients can be
transfused with D positive RBCs. Thus, with few exceptions, from a historical
perspective, one can safely classify the weak D as D positive.
This question gets a little trickier
when dealing with females of childbearing age. We particularly want to avoid
giving Rh positive blood to females to avoid anti-D and the complications of
Hemolytic Disease of the Fetus and Newborn. Therefore, when dealing with these
patients, lab policies and physicians tend to be more conservative in their
approach to transfusion. The consequences, however, in males and older females
are less serious and these patients could be given Rh positive blood if there
exists a shortage of Rh negative units. Any patient who becomes alloimmunized
to the D antigen, would thereafter be transfused with Rh negative products.
Does an “O
Du” patient need to receive RhoGAM if she pregnant and her husband is Rh
positive?
This, again, would be up to the medical
director, the lab’s SOPs or the patient’s physician. Depending on lab practice,
the lab may or may not perform weak D testing. If the lab does not perform weak
D and results this patient as Rh neg, the patient would get Rhogam. If the lab
does do weak D testing and finds a weak D phenotype, the decision whether or
not to give Rhogam would be up to lab practices and the practitioners involved.
The lab’s policy on terminology used in resulting the type may also reflect the
decision whether or not to give Rhogam. This brings up a lot of questions in
the lab because we know that a patient who would not make anti-D would not need
Rhogam. So, what is the best course of action? Read my next blog to learn more
about troubleshooting and resolving D typing discrepancies!
From the discrepancies in reported type in this individual, and putting all the pieces of the puzzle together, we can conclude that this patient is a weak D phenotype. However, the type reported and the terminology used varies from lab to lab. Molecular testing is available, yet most labs are still using serological testing for blood types for both donors and patients. This is based on several factors within the lab setting. Stay tuned for my next Blood Bank blog exploring D typing discrepancies and the financial aspects of performing genotype on pregnant patients to clarify Rh type.
-Becky Socha, MS, MLS(ASCP)CM BB CM graduated
from Merrimack College in N. Andover, Massachusetts with a BS in
Medical Technology and completed her MS in Clinical Laboratory Sciences
at the University of Massachusetts, Lowell. She has worked as a Medical
Technologist for over 30 years. She’s worked in all areas of the
clinical laboratory, but has a special interest in Hematology and Blood
Banking. When she’s not busy being a mad scientist, she can be found
outside riding her bicycle.
A 28 year old
woman, gravida 1 para 0 presented to her OB/GYN for her first prenatal visit. A
type and screen was ordered and the patient typed as A pos, with a positive
antibody screen. Maternal history indicated that she had received several transfusions,
for a total of 5 units of blood, following an automobile accident 15 months
previously. An antibody identification was performed and Anti-K was identified
in her plasma. The patient sample was phenotyped and was confirmed to be K
negative.
Is the fetus at
risk for Hemolytic Disease of the Fetus and Newborn (HDFN)? How can we know? And,
if so, how should this pregnancy be monitored?
The answer to
these questions is not a simple answer, and depends on several factors. Let’s
look first at HDFN and its causes. HDFN is destruction of the RBC’s of the
fetus and newborn by antibodies produced by the mother. This happens in 2
steps. The first step is that blood containing
a foreign antigen enters the maternal blood stream and stimulates the mother to
produce unexpected IgG antibody. But, how is a mother exposed to these foreign
antigens? The mother is exposed either via a blood transfusion or a previous
pregnancy. In this case, this was the mother’s first pregnancy, however her
history revealed that she had been previously transfused. In order for the
mother to produce anti-K , she must be K antigen negative, which was confirmed
in the Blood Bank testing. She was exposed to the K antigen through transfusion
and produced the anti-K antibody to the foreign antigen. The second step in the
development of HDFN occurs when the
mother’s antibody crosses the placenta and binds
to this foreign antigen
present on the red blood cells of the fetus. This can lead to RBC
suppression, destruction, and fetal anemia.
Again, certain
criteria must be met. First of all, the antibody must be IgG. Only IgG
antibodies can cross the placenta. Active transport of IgG from mother to fetus
begins in the second trimester and continues until birth. Secondly, the mother’s
antibody is only of concern if the baby possesses the antigen that the mother
lacks. Where does the baby get an antigen
that is foreign to the Mom??
It’s the Dad’s Fault!! In HDFN, the mother lacks the antigen in question and the
fetus possesses the antigen, which is of paternal origin.
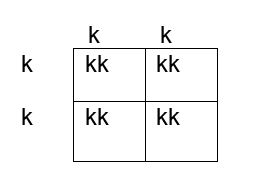
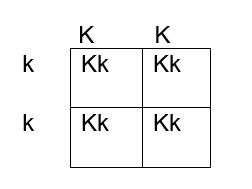
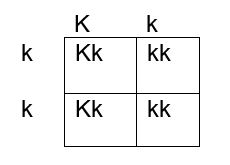
How do we determine if the fetus has the K antigen and is at risk? If you remember your genetics and Punnett squares, if the mother does not have the antigen and the baby does, the father must possess the antigen, because the baby gets an allele from each parent. This means that the fetus affected by HDFN is always heterozygous for the antigen in question. Figures 1, 2 and 3 below illustrate the possible inheritance patterns. In the first scenario, shown in Figure 1, the baby would not inherit a K antigen and would not be at risk for HDFN. In the Figure 2 scenario, the father is homozygous for K, and 100% of offspring from these parents would be K positive. Figure 3 illustrates a heterozygous father who would have a 50% chance of passing this gene to their offspring.
Figure 1. Punnett square showing inheritance of K antigen. Mother (on side) is negative for K (kk), father (at top) is also negative, homozygous kk
Figure 2. Punnett square showing inheritance of K antigen. Mother is negative for K (kk), father is homozygous KK
Figure 3. Punnett square showing inheritance of K antigen. Mother is negative for K, father is heterozygous Kk
The father was phenotyped as K positive. The father’s blood sample was sent out for further zygosity testing, and he was found to be heterozygous for the K antigen. Thus, the fetus had a 50% chance of being affected by HDFN, and further testing was performed. The mother’s antibody titer was 1:4. To avoid an invasive procedure such as amniocentesis or chorionic villus sampling (CVS) which may worsen maternal alloimmunization, fetal DNA was isolated from the mother’s plasma at 12 weeks’ gestation and the fetal genotype was determined. The fetus was determined to be K positive and at risk for HDFN.
The mother’s
titer and the fetus continued to be monitored. Diagnostic ultrasounds were performed to monitor fetal size,
age, and structural changes. At
16-18 weeks’ gestation, ultrasounds of the middle cerebral artery (MCA-PSV) were
performed to assess fetal anemia. MCV-PCA of 1.29 -1.5 multiples of mean (MoM)
for the gestational age is indicative of mild anemia. Higher values predict
moderate to severe anemia which require further intervention. At 18 weeks the
MCV-PCA was 1.27 MoM and the fetus was determined to be developing normally.
A type and screen and antibody titer at 28 weeks
showed the mother’s titer had increased, to 1:32, indicating that fetal RBCs
with K antigen had entered the mother’s circulation and were stimulating further
antibody production. Repeat MCV-PCA was 1.33, indicating mild anemia. Weekly
measurements of MCA-PCV were recommended. At 32 weeks, a sudden increase was
recorded, with MCV-PCA of 1.65 MoM. Cordocentesis was performed and fetal
hemoglobin was 6.2g/dl. Fetal DAT was positive and anti K was identified in the
eluate. An intrauterine transfusion (IUT) was performed. IUT was repeated at 34
and 36 weeks. The infant was delivered at 37weeks. The newborn required several
neonatal transfusions while in the hospital and was discharged to home 3 weeks
later.
Kell isoimmunization is the third most common cause of HDN
after Rh and ABO and the most clinically significant of the non-Rh system
antibodies in the ability to cause HDFN.
It tends
to occur in mothers who have had several blood transfusions in the past, but it
may also occur in mothers who have been sensitized to the K antigen during
previous pregnancies. Anti-K HDFN may cause rapidly developing severe fetal
anemia. Anemia and hypoproteinemia are dangerous to the unborn child because
they can lead to cardiac failure and edema, a condition known as hydrops
fetalis. The MCA-PSV is a non-invasive doppler measurement of peak systolic
velocity which is used to monitor fetal anemia. As mentioned previously,
MCV-PCA of 1.29 -1.5 multiples of mean (MoM) is indicative of mild anemia. Values
greater than 1.5 MoM are very sensitive and can be used to predict moderate to
severe anemia that would need intervention.
HDFN due to anti-K differs from ABO and Rh HDFN in that, in
HDFN due to K alloimmunization, Anti-K targets the RBC precursors. Remember
that the K antigen can be detected on fetal RBCs as early as 10 weeks. The primary mechanism of K HDFN is due to maternal
anti-K antibody actually
suppressing the fetal production of RBCs, rather
than hemolysis of mature fetal RBCS as seen in ABO and Rh HDFN. With reduced
hemolysis, amniotic fluid bilirubin levels also do not correlate well with the degree
of anemia. In addition, alloimmunization due to Anti-K differs in that even a
relatively low maternal anti-K titer can cause erythropoietic suppression and
severe anemia. In Rh HDFN, a critical titer is considered to be 16. In anti-K
HDFN, a critical titer is considered to be 8, and newer research suggests a titer of 4 should be used to target clinical monitoring.4 Since fetal anemia
can occur even with low titers, and the titer does not necessarily correlate to
the degree of anemia, fetal MCA-PSV measured by Doppler ultrasound is the
investigation of choice in the evaluation of anemia related to maternal K alloimmunization.
-Becky Socha, MS, MLS(ASCP)CM BB CM graduated from Merrimack College in N. Andover, Massachusetts with a BS in Medical Technology and completed her MS in Clinical Laboratory Sciences at the University of Massachusetts, Lowell. She has worked as a Medical Technologist for over 30 years. She’s worked in all areas of the clinical laboratory, but has a special interest in Hematology and Blood Banking. When she’s not busy being a mad scientist, she can be found outside riding her bicycle.
The patient is a 54 year old woman, presenting to the
Emergency Room with complaints of abdominal cramps and feeling lethargic for
the past few days. She also reports her stools have been black and sticky. Her chart reveals a history of ulcers and GI
bleeding. She was transfused with 2
units packed RBCs 2 months ago for the same symptoms. CBC results are shown
below.
The patient was admitted to the hospital and four units of
blood were ordered. The patient is type A pos with a negative antibody screen.
One unit of packed red blood cells would be expected to raise the Hgb by 1g/dl.
Because the patient was actively bleeding, 4 units were crossmatched and
transfused.
Two days later, the patient was discharged, with orders to
follow up with her GI doctor for further testing and treatment. Three days
after discharge she still felt weak and returned to the ER. On examination, it
was noted that the patient’s eyes and skin appeared jaundiced. The patient had
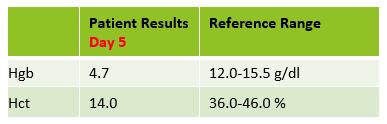
a fever of 100F. Repeat lab results are shown below.
The Physician ordered a type and crossmatch for 2 units of
packed red blood cells. The patient’s antibody screen was now positive. A
transfusion reaction workup was initiated
Transfusion workup
Clerical Check- No clerical errors found.
Segments from all 4 transfused units were phenotyped for Jka
antigen. Three of the four units transfused typed as Jka positive.
A transfusion reaction is defined as any transfusion-related adverse event that occurs during or after transfusion of whole blood, or blood components. Transfusion reactions can be classified by time interval between the transfusion and reaction, as immune or non-immune, by presentation with fever or without fever, or as infectious or non-infectious.
A delayed
transfusion reaction is defined as one whose signs or symptoms typically present
days to several weeks after a transfusion. In Transfusion Medicine, we do not
want to give the patient an antigen that is not present on their red blood
cells. However, we do not routinely phenotype patients, so, in the patient with
a negative antibody screen and history, it is always possible that the patient
receives units with foreign antigens. The more immunogenic the antigen, and the
greater number units received that expose the patient to this antigen, the
greater likelihood that the patient will develop an antibody to the foreign
antigen. Therefore, this type of reaction would also be categorized as immune.
In a delayed hemolytic transfusion reaction (DHTR)
investigation, the units transfused would have appeared compatible at initial
testing. This type of adverse event is fairly common in patients who have been
immunized to a foreign antigen from previous transfusion or pregnancy. The antibody formed may fall to a very low level
and therefore not be detected during pretransfusion screening. If the patient
is subsequently transfused with another red cell unit that expresses the same
antigen, an anamnestic response may occur.
The antibody level rises quickly and leads to the DHTR. In the transfusion reaction workup,
this antibody can often be detected when testing is repeated. However,
in some cases, particularly with Kidd antibodies, the levels again drop off so
quickly they may not be detected! The diagnosis of DHTR is often difficult because
antibodies against the transfused RBCs are often undetectable and symptoms are
inconclusive.
This case is a classical example of a DHTR. Kidd antigens are notorious for causing DHT
because their levels can drop off quickly and disappear, making them difficult
to detect in screening. In this case, the transfusion two months earlier
exposed the patient to the Jka antigen and the patient produced the
corresponding antibody. The levels then dropped quickly, as elusive Kidds are
known to do! When the patient returned to the ER in crisis, the antibody levels
had dropped below detectable levels and the antibody screen was negative. The
patient was given 4 units and returned to the ER five days after transfusion. This
patient did exhibit mild jaundice and a low-grade fever. However, often, the
only symptom of a DHTR is the unexpected drop in Hgb and Hct, making them even
more difficult to diagnose.
The new antibody screen, sent to the Blood Bank on day 5, detected
anti-Jka. The DAT was positive mixed field due to the transfused cells. Elution
was performed and anti-Jka was recovered in the eluate. In the DHTR, only the
transfused cells are destroyed. Phenotyping segments from the transfused units
can estimate amount of transfused RBCs that may have shortened survival. Management
of this case patient would be to provide antigen negative units for all future
transfusions.
Kidd (Anti-Jka
and Anti-Jkb), Rh, Fy, and K have all been associated with DHTR and
occur in patients previously immunized to foreign antigens through pregnancy
and transfusion. These types of reactions are generally self-limiting but can
be life threatening, especially in multiply transfused patients, such as those
with sickle cell anemia. Antigen negative blood must always be given, even if
the current sample is not demonstrating the antibody in question. For that
reason, it is vitally important to always do a thorough Blood Bank history
check on all samples!
-Becky Socha, MS, MLS(ASCP)CM BB CM graduated
from Merrimack College in N. Andover, Massachusetts with a BS in
Medical Technology and completed her MS in Clinical Laboratory Sciences
at the University of Massachusetts, Lowell. She has worked as a Medical
Technologist for over 30 years. She’s worked in all areas of the
clinical laboratory, but has a special interest in Hematology and Blood
Banking. When she’s not busy being a mad scientist, she can be found
outside riding her bicycle.
In the Iliad, Homer described the chimera as “a thing of immortal make, not human, lion-fronted and snake behind, a goat in the middle, and snorting out the breath of the terrible flame of bright fire (1).” This mythical creature has a lion’s head, a goat’s middle and the tail of a serpent, and the siting of a chimera was considered to be an omen for disaster! Thankfully, not so much in blood bank. Though ABO discrepancies can be a challenge, even most chimeras can easily be resolved with a few additional steps and a patient history.
Figure 1. The mythical chimera.
To review, an ABO discrepancy occurs when unexpected
reactions occur in the forward or reverse grouping, or the forward typing does
not match the reverse typing. Some weak subgroups of A (notably A3)
are known for giving mixed field reactions. Weak activity with anti-A, anti-B or
anti-D can also in result mixed field reactions in leukemia patients. In these examples,
the mixed field reactions are due to the weakened expression of the
corresponding antigens.
Chimerism is the presence of 2 cell populations in a single
individual. There are scenarios
where ABO discrepancies causing mixed field reactions indicate an apparent
chimera. A group A positive patient who received several units of O
negative blood will have mixed field reactions due to the presence of two blood
types in their peripheral blood. This would be a temporary situation. A patient
who received a bone marrow or stem cell transplant from a non-group identical
donor will have 2 populations of red blood cells until the new type is
established. We refer to these as artificial chimera cases, as the second blood
type is not naturally occurring, but present due to the introduction of a
different blood type via transfusion or transplantation.
Table 1. Group A pos patient who received several units of group O neg red cells
Like the
mythical beast, a chimera in biology describes an organism that has
cells from two or more zygotes. When chimerism exhibits only in the blood, the phenomenon
can be termed an artificial chimerism, as described above, as dispermic
chimerism or as twin chimerism. Dispermic chimerism occurs in other animal
species but is a rarity in humans. It occurs when 2 eggs are fertilized by 2
sperm and these products are fused into one body. In this case, the chimerism
is not limited to blood, but may also result in hermaphroditism, or two
different skin colors or eye colors.
Twin chimerism occurs when, in utero, one twin transfuses
blood cells, including stem cells, to
the other. Sine the fetal immune system is immature, the host does not see
these transfused blood cells as foreign antigens. The stem cells can proliferate and this
results in the production of cells from both the donor and the host for the
rest of the individual’s life. Two non-compatible blood groups can co-exist in
one individual! This phenomenon is usually discovered by coincidence during a
routine type and screen. This patient could be found to have mixed field or
weak reactions on ABO typing, or could have missing reactions in the back type,
all with no history of transfusion, transplantation and no disorder that could
explain the findings. What is a tech to do? An important step in resolving all
ABO discrepancies is to review patient history.
In 1953 a human chimera was reported in the British Medical Journal. A woman was found to have blood containing two different blood types. Apparently this resulted from her twin brother’s cells living in her body (2). More recently, in 2014, a case described in Blood Transfusion describes a 70 year old female who was found to have mixed field reactions with ABO and RhD typing during routine testing before surgery. She had no history of transfusion or transplantation, and a history of seven pregnancies. Repeat testing by other methods and with different reagents gave the same results. On further questioning, the patient affirmed that she had been born a twin, but her twin brother had died as an infant. Since chimerism was suspected, molecular typing and flow cytometry were performed. The presence of male DNA was found by PCR testing and flow cytometry confirmed two distinct populations of red blood cells (3).
Twin chimeras with mixed blood types of 50%/50% or 75%/25%
are easily picked up in ABO typing as mixed field reactions. A twin chimera with
95% group O blood and 5% group A may show a front type of a group O and a back
type that lacks anti-A . Because there is immune tolerance to A cells from the
twin, the expected naturally occurring anti-A is not present. On the other
hand, a twin chimera who is primarily group A with 5% O cells would not be
recognized as a chimera in routine ABO typing.
Table 2. Group O chimera with 5% minor cell population A cellsTable 3. Group A chimera with 5% O cells
How common is blood group chimerism? A 1996 study found that such blood group chimerism is not rare. Though we do not often encounter this in blood bank, their study of 600 twin pairs and 24 triplet pairs showed that this occurs more often than was originally thought, with a higher incidence in triplets than in twins. Because it does not cause any symptoms or medical issues, many such chimeras go undetected. In addition, the study found that many of these chimeras had very minor second populations, making them undetectable in serological testing. In blood bank, we generally test for ABO/RH and do not test for other antigens in routine testing. The study used 849 marker antigens. They also used a very sensitive fluorescent technique which they developed for detecting these very subtle minor populations. This study showed that while chimeras are not rare, they are something that, with present testing methods, we will not encounter too often (4).
Dual cell populations induced by chimeras have been the subject
of many studies. Historically, most chimeras were naturally occurring. With
newer medical interventions and therapies, we may see more situations that lead
to mixed cell populations. Transfusion, stem cell transplants, kidney
transplantation, IVF and artificial insemination can all lead to temporary and
sometimes permanent chimeras. These can present challenges in the blood bank
laboratory in interpreting results and for patient management. A question of
chimera presentation can usually be solved by putting on our detective hats and
investigating patient history. Further testing can be done with flow cytometry
and molecular methods, if needed. Modern medicine may have given us more blood
bank challenges but modern technology has equipped us with newer methods to
solve them. A chimera is no longer a sign of impending trouble!
References
Homer, Iliad. In Richmond Lattimore’s Translation.
Bowley, C. C.; Ann M. Hutchison; Joan S. Thompson; Ruth Sanger (July 11, 1953).“A human blood-group chimera” (PDF).British Medical Journal: 8
Sharpe, C.; Lane, D.; Cote J.; Hosseini-Maaf, B.; Goldman, M; Olsson, M.; Hull, A. (2014 Oct ). “Mixed Field reactions in ABO and Rh typing chimerism likely resulting from twin hematopoiesis”, Blood Transfusion:12(4): 608-610
-Becky Socha, MS, MLS(ASCP)CM BB CM graduated from Merrimack College in N. Andover, Massachusetts with a BS in Medical Technology and completed her MS in Clinical Laboratory Sciences at the University of Massachusetts, Lowell. She has worked as a Medical Technologist for over 30 years. She’s worked in all areas of the clinical laboratory, but has a special interest in Hematology and Blood Banking. When she’s not busy being a mad scientist, she can be found outside riding her bicycle.