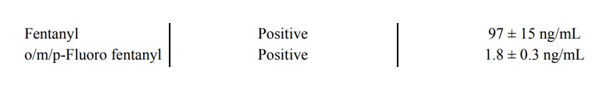When the Medical Examiner’s office receives a report of someone dead in a bathtub, the reporting party often presumes that the individual must have drowned. Yet deaths in bathtubs can be surprisingly complex. We initially discussed drowning back in June of 2023 but to summarize, drowning is a diagnosis of exclusion. The few signs of drowning at autopsy (pulmonary edema, watery fluid in the stomach and sinuses), are neither sensitive nor specific, so it is critical to exclude other potential causes of death. Particularly when a bathtub (or hot tub) is involved, a methodical step-by-step approach is helpful to avoid jumping to inaccurate conclusions.
The first step for us is to determine if the tub contained water. While this may sound obvious, this critical detail isn’t always reported initially. The first person on scene often reflexively turns off running water and opens the drain – and (understandably) in the heat of the moment, this alteration of the scene might not be documented. Our investigators are experienced at identifying indirect evidence of a wet bathtub, such as a water line, wet clothing, or wet sponges and washcloths.
Simply having water in the tub isn’t sufficient, though. We need to know whether the person’s nose and mouth were beneath the water. Sometimes, the physical dimensions of the tub and the position in which the person was found are incompatible with complete submersion. If the body has been moved, though, this evidence may be lost. Conversely, just because someone’s face is under the water doesn’t prove that they drowned; someone may die suddenly from heart disease while filling the tub, for example, and subsequently the water rises.
As mentioned earlier, there are no pathognomonic signs of drowning at autopsy. This isn’t necessarily a problem in the proper context. Take this example: a person who doesn’t know how to swim (and isn’t wearing a flotation device), falls out of a boat into a natural body of water. The body is recovered a day later. An autopsy reveals no “typical” drowning findings, but no other potential causes of death. In this scenario, drowning is the only remaining reasonable option for a cause of death. But to diagnose drowning in a shallow body of water, we need to explain why the person couldn’t escape the environment. In other words, in a bathtub, why didn’t the decedent just sit up? Sometimes, the reason is the age of the deceased – infants or young toddlers clearly can’t escape a deep bathtub, but even large buckets can be dangerous if they fall head-first (figure 1). Elderly and frail individuals, or those with neuromuscular conditions, may be unable to pull themselves up to a sitting position. Conditions like epilepsy can be dangerous if a seizure occurs while the person is bathing, and intoxication by alcohol, opiates, or other sedative-hypnotic drugs may cause someone to lose consciousness and slip beneath the water.
As you can see, there are a lot of avenues for interrogation investigating a death in a bathtub. That’s why these cases can be excellent examples of forensic casework – the correct answer is only identified following thorough scene investigation, autopsy, toxicology, and a review of the medical history.
Figure 1: This illustration released by the U.S. Consumer Product Safety Commission warns that 5-gallon buckets, in particular, are a drowning hazard for young children.
We’ve previously addressed the basics of gunshot wounds (see https://labmedicineblog.com/2023/09/22/the-ins-and-outs-of-gunshot-wounds/) but forensic pathologists need to be familiar with injuries inflicted by a variety of firearms. If you grew up in a rural area (like me) you are probably familiar with shotguns as a typical hunting tool. However, shotguns also have several unique characteristics which are crucial for forensic pathologists to understand.
First, the barrel of a shotgun is most often a “smooth bore” as opposed to the longitudinal spiraling lands and grooves (or “rifling”) found in the barrels of rifles and handguns. This means traditional ballistic “matching” (testing to see if a bullet was fired from a particular weapon) is impossible.
Secondly, shotgun ammunition is constructed differently. Broadly speaking there are three types of shotgun ammunition – birdshot, buckshot, and slugs (from smallest to largest in individual size).
A single shotgun projectile is composed of the ‘shell’, an outer casing of plastic with a metal base. The shell contains primer and gunpowder, “wadding” (fiber or cardboard material), and a plastic “sleeve” which holds the projectile(s) (or “shot”). The individual characteristics can vary depending on the “gauge” (caliber) used, but a single shell will typically contain hundreds of birdshot pellets, tens of buckshot pellets, or one slug. The sleeve initially holds these individual pellets together – but upon leaving the barrel, the plastic flays outward, and the pellets or slug are released. For birdshot and buckshot, this means the individual pellets begin to spread apart and lose speed.
This spread of pellets explains why shotguns are a popular choice for hunting birds – a small, constantly moving target. It also helps us determine the range of fire from the shape of the entrance wound. At a close or contact range, there is minimal opportunity for the pellets to spread, resulting in a single circular wound. As the distance from the target increases to several feet, the wound edges become scalloped and individual pellet wounds are observed around the main entrance wound. At a distant range (approximately ten feet), there are only individual pellets wounds. Importantly, these wound characteristics can only be assessed on the skin surface – not on radiographs, which will only show the pellets in their final location within the body.
The other components of the shell can add to the wound characteristics. At contact range, the plastic sleeve (and even the shell) will enter the body but will likely not be visible by radiograph – these still need to be recovered as evidence. At medium distances, the sleeve and/or shell may strike the skin surface and impart a distinct patterned abrasion, without penetrating the skin.
Fortunately, shotgun wounds are a less common part of day-to-day practice – yet it is still important to be prepared with a basic understanding of how these weapons function and the diverse types of ammunition available.
The petri dish on the left holds a representative sample of birdshot pellets, recovered from a contact-range shotgun wound. The plastic sleeve (right) was also in the wound – note the opened flaps.This contact-range gunshot wound is large and circular, although there is still some faint scalloping of the edges. The black discoloration to the left is caused by searing of the skin from the hot gas exiting the shotgun barrel.This intermediate-range shotgun wound has a central main wound with scalloped edges and surrounding satellite entrance wounds caused by the pellets beginning to spread.At distant range, all the pellets have dispersed. At this range the pellets have lost energy, and the wounds are often superficial; however, depending on the location of the injury on the body, even single pellets can cause lethal trauma.The dispersal of individual pellets within the body can lead to unexpected findings. In this autopsy, the decedent had a self-inflicted shotgun wound to the chest with birdshot. A few pellets entered the aorta near the arch, and several embolized down the length of the aorta before lodging in the right iliac artery (shown here). This illustrates why the spread of pellets should only be assessed on the skin surface, and not based on radiographs.
References
DiMaio, Vincent J. Gunshot Wounds. 3rd ed., CRC Press, Taylor & Francis Group, 2016.
Dolinak, et al. Forensic Pathology: Principles and Practice. Elsevier Academic Press, 2005.
-Alison Krywanczyk, MD, FASCP, is currently a Deputy Medical Examiner at the Cuyahoga County Medical Examiner’s Office.
Let’s imagine you are a forensic pathologist, called by investigators to the basement of an abandoned house where a building inspector found human remains. Upon your arrival, you identify a human skeleton, still partially encased in trash bags. The plastic trash bags have melted over the exposed surfaces, and there are charred cans of lighter fluid lying on top of the body. After a full examination of the body at your morgue, however, you cannot find any remaining signs of injury. One need not be an expert to recognize that the circumstances in which this body was found are extremely concerning, regardless of the absence of injuries at autopsy. This is when the diagnosis of “homicide by unspecified means” (“HUM”) enters consideration.
As we have previously discussed (see Undetermined, Undetermined – Lablogatory), in situations of extensive soft tissue loss or incomplete remains, we may not be able to identify the cause of death. It is unfortunately not uncommon for bodies of homicide victims to be concealed, delaying their discovery and allowing decomposition to progress. Out of a desire to hide the victim’s identity or to conceal the crime itself, attempts may be made at dismembering or destroying the body, which can further hinder efforts to identify injuries. Still, there are situations which are easily recognized as suspicious – yet we need to be careful to not rush to conclusions. There are less malignant explanations for some strange circumstances – for example, a person who accidentally overdoses may be moved to a different location, to divert law enforcement attention from a specific house or person. A hiker in an isolated location may suffer a natural cardiac event but not be found before decomposition and animal predation have occurred.
There are five criteria required to meet the diagnosis of “homicide by unspecified means,” a term which was originally coined by the late Dr. Joe Davis in Miami, Florida. These criteria were designed to ensure that all possible alternatives are thoroughly considered before arriving at the diagnosis. The criteria, as delineated in the original 2010 article, are:
Objectively suspicious circumstances of death. This would include evidence that the body was deliberately concealed (in trash bags, luggage, or a shallow grave, for example), attempts were made to destroy the body (e.g. with fire, or bleach), or that the victim was restrained. There may be evidence at the scene (or in the victim’s home) of significant blood loss, or there may still be non-lethal injuries (for example, a shallow laceration, or numerous bruises) identifiable on the body.
No anatomic cause of death. Meeting this criterion requires the completion of a full autopsy, despite the potential lack of tissue or complete remains. Sometimes evidence of lethal trauma is still identifiable – for example, gunshot wounds or knife marks on bone – and in this situation, the better “cause of death” is the specific type of injury. In other situations, there may be significant cardiovascular disease found at autopsy; the presence of a competing, natural cause of death must be carefully weighed with the other evidence.
No toxicological cause of death. This essentially means we have excluded an overdose as a possible cause of death. At times, this is a difficult standard to meet – in the example of skeletal remains, no material may be present to test. Other remains may be so decomposed that obtaining quantitative results (rather than qualitative) is impossible. Even if results are ‘positive’ for drugs of abuse, there is strong evidence that intoxicated individuals are at increased risk for interpersonal violence, and it would be a public disservice to automatically ascribe these deaths to overdose. This criteria, much like criteria #2, needs to be considered carefully in the context of all other findings.
No environmental, circumstantial, or historical causes of death. One always needs to consider the scene investigation and surrounding environment. Take the example above of a hiker in an isolated location – a death from hypothermia can have no or minimal findings at autopsy, let alone in the context of decomposition. Deaths due to drowning, epileptic seizures, or transient exposure to a toxic agent (like carbon monoxide) are similarly difficult to identify in these circumstances. If any of these possibilities cannot be confidently excluded, the diagnosis of “HUM” should not be made.
A more specific cause of death cannot be suggested by the data set. Essentially a reminder to review the totality of the evidence. Ruling a death as a ‘homicide’ sets off a chain of events which could result in a person being permanently incarcerated or executed. It is not a ruling to be made lightly.
Not every jurisdiction uses “HUM” as a term. Some will rule the cause of death as purely ‘undetermined’ and the manner of death ‘homicide’. In either case, the point is the same – there are clear indicators of homicidal violence, yet we cannot determine the specific type – and these criteria are still helpful to make sure alternative manners of death are thoroughly considered.
References:
Matshes EW, Lew EO. Homicide by unspecified means. Am J Forensic Med Pathol. 2010 Jun;31(2):174-7. doi: 10.1097/PAF.0b013e3181df62da. PMID: 20436340.
Krywanczyk A, Gilson T. Homicide by Unspecified Means: Cleveland 2008 to 2019. Am J Forensic Med Pathol. 2021 Sep 1;42(3):211-215. doi: 10.1097/PAF.0000000000000657. PMID: 33491950.
-Alison Krywanczyk, MD, FASCP, is currently a Deputy Medical Examiner at the Cuyahoga County Medical Examiner’s Office.
In several previous blogs, I’ve mentioned the topic of post-mortem radiography (or “x-rays”). While postmortem CT scanning is a hot topic in the field, plain films are a tool which has been in widespread use for decades. Autopsy standards of the National Association of Medical Examiners require, at a minimum, radiographs be performed on all infants, gunshot wound victims, explosion victims, and charred or decomposed remains. Let’s examine the reasons for these requirements and look at a few specific examples.
All infants must get full body x-rays to check for acute or healing rib and long bone (extremity) fractures, which could be indicative of physical abuse. The extremities are not usually dissected in the course of a typical post-mortem examination, but fractures that can’t be attributed to birth trauma (especially in a pre-mobile child) are a concerning finding that needs further investigation and dissection.
Radiographs taken of gunshot wound victims document the presence and location of retained projectiles, all of which need to be recovered as evidence.
This x-ray of a gunshot wound victim shows two separate types of ammunition, seen as radio-opaque (white) in the image. The smaller, circular pieces are “birdshot” shotgun ammunition (blue arrows) while the larger pieces are traditional handgun ammunition (red arrow).
Radiographs are commonly performed in any type of penetrating trauma, including sharp force injury and explosive injuries, to identify retained foreign bodies (especially broken fragments of the blade, which may pose a risk to the pathologist). Similar to projectiles, these fragments need to be recovered as evidence. Radiographs can also document the presence of an air embolism, which can be missed at autopsy if special dissection techniques aren’t performed.
This individual had sharp force injuries to their neck, which injured large veins. The x-ray in this case was performed to see if any fragments of the weapon were still in the body, but also showed a large air embolism in the right atrium and ventricle (blue arrows), seen as radiolucency (gray/black). Open injuries to veins in the head and neck can cause air emboli as breathing creates negative intra-thoracic pressure, drawing air inward though any open channels.
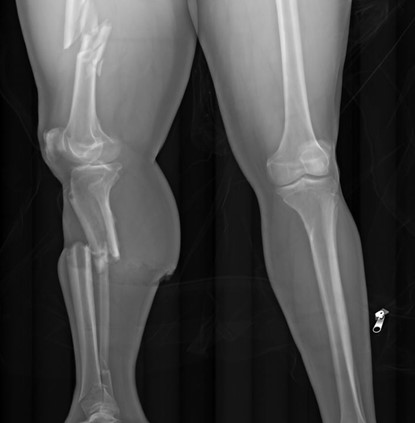
In pedestrians who have been struck by motor vehicles, radiography is the first step in examining trauma. As mentioned earlier, the extremities aren’t typically dissected during a traditional autopsy – but in pedestrians, lower extremity fractures can document the site of initial impact, and the distance of the fracture from the foot may indicate the bumper height of the car.
This x-ray of a pedestrian struck by a motor vehicle shows displaced fractures of the right femur, tibia, and fibula. Note the body bag zipper in the lower right corner.
Fire victims may have extensive thermal injuries and charring which can hide evidence of other injuries, and radiographs can help identify them. Radiographs are also one step toward identifying the victim. Decomposed bodies must be x-rayed for similar reasons – external and internal soft tissue alterations make assessment for trauma more difficult, and radiographs may be needed to confidently identify the body.
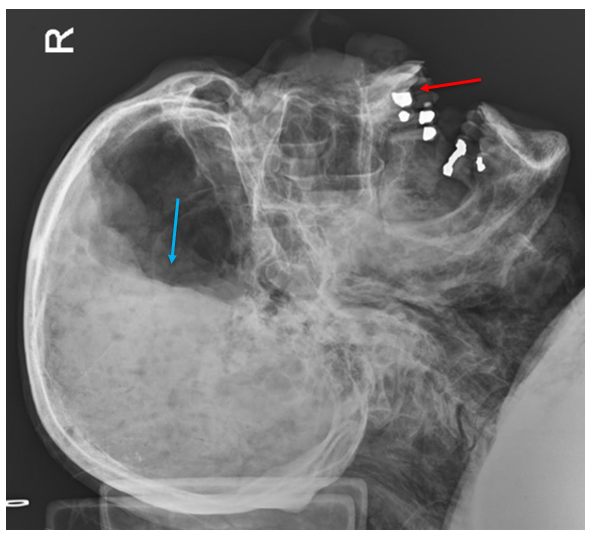
In this x-ray of a decomposed person, dental fillings are easily visible (red arrow), which are typically unique for an individual and can be used for identification. Note the irregular shapes of the fillings, which are just as important as their location. Also note the air-fluid level in the cranial cavity (blue arrow), indicating complete liquefication of the brain.
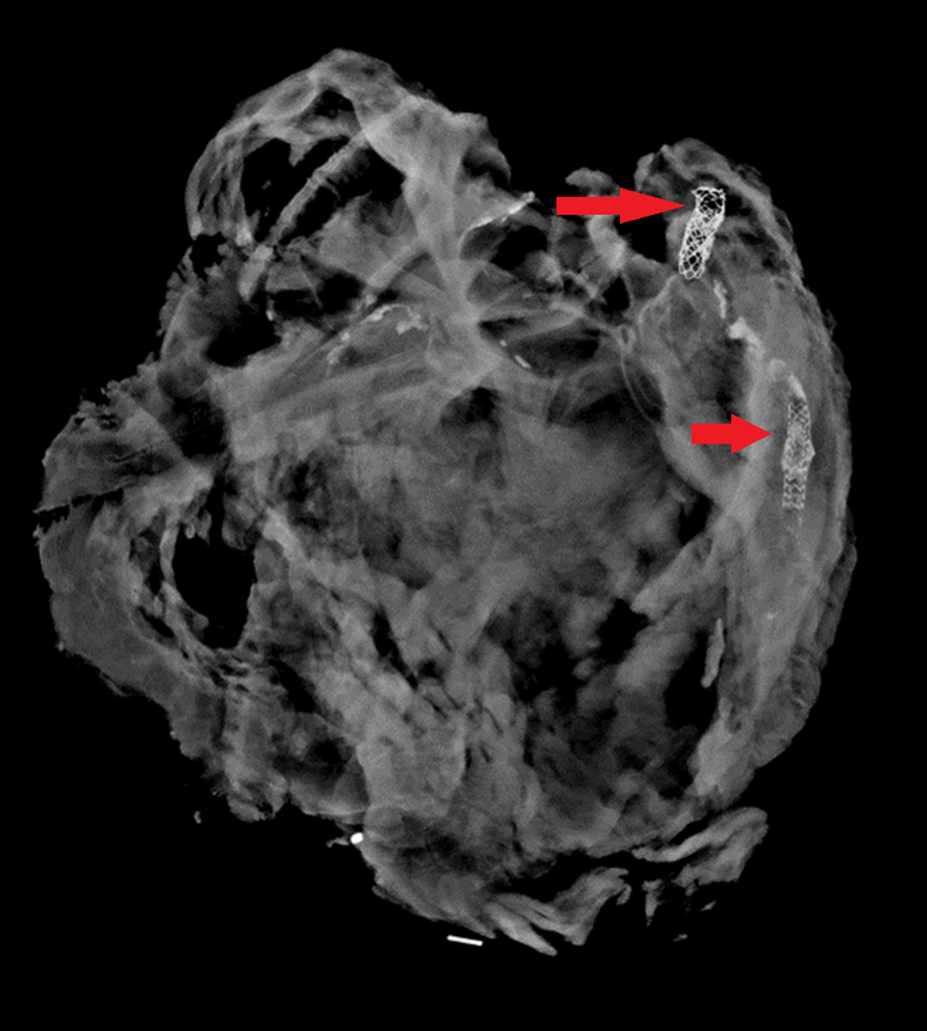
On occasion, radiographs can be performed on individual organs to help define anatomy or previous surgical alterations. Especially in the heart, radiographs can demonstrate coronary artery or valve calcifications, surgical clips from bypass grafts, or other radiopaque prostheses. Knowing the location of devices before dissection gives the pathologist a better chance at preserving and evaluating important structures.
This heart was x-rayed after removal, as the decedent had a history of heart disease with prior interventions. Coronary stents (red arrows) are easily visible; in practice, they can be difficult to find on gross dissection in patients with heavily calcified arteries.
Not all offices can afford the installation (or maintenance) of CT scanners, but access to x-ray machines is more widespread. As we’ve seen here, x-rays are a versatile tool which can document injuries, help identify decedents, and direct the pathologist to perform special autopsy procedures which aren’t part of the daily routine.
-Alison Krywanczyk, MD, FASCP, is currently a Deputy Medical Examiner at the Cuyahoga County Medical Examiner’s Office.
A recent report from the Centers for Disease Control and Prevention (CDC) revealed that after the emergence of the COVID-19 pandemic, antibiotic resistance has increased by at least 15%. In particularly, the extended spectrum beta-lactamase (ESBLs) producing Enterobacterales have gone up to 32%. This group includes E. coli, K. pneumoniae, K. oxytoca and P. mirabilis.1
ESBLs are β-lactamases capable of conferring resistance to β-lactam antibiotics such as penicillins, first, second and third generation cephalosporins and aztreonam (but not cephamycins or carbapenems) (Table 1). A key characteristic is that these enzymes hydrolyze the antibiotics but are inhibited by β -lactamase inhibitors such as clavulanic acid, tazobactam and sulbactam. Thus, diagnostic assays utilize this key characteristic to develop tools to screen for ESBL producers as discussed below.1-3
Figure 1. Disk diffusion test for ESBL detection. (A) A >5mm difference between the cefotaxime (CTX) and cefotaxime/clavulanic acid (CTX/CLA) disks, and ceftazidime (CAZ) and ceftazidime/clavulanic acid (CAZ/CLA) disks suggest a ESBL producer (top). (B) A <5 mm difference between CTX and CTX/CLA disks, and CAZ and CAZ/CLA does not suggest an ESBL-producer.
The genes encoding ESBLs are found on plasmids which enable rapid and easy transfer between Enterobacterales and some non-Enterobacterales as well. TEM-1, one of the first plasmid encoded β-lactamase enzymes, was identified in E. coli. Another one, SHV1 was subsequently discovered in Klebsiella.2 These original β-lactamases were narrow spectrum, but mutations have led to enzymes with broad spectrum activity, hydrolyzing many of the commonly used antibiotics. Today, there are over 100 variations of these enzymes that have spread resistance worldwide. The most dominant ESBL today is CTX-M, an enzyme that originated from Kluyvera species. CTX-M encoded resistance has now been reported among Enterobacterales as well as P. aeruginosa and Acinetobacter sp.2 Other ESBL families include IRT, CMT, GES, PER, VEB, BEL, TLA, SFO, and OXY.4
Detection of ESBL producers
The most common method for ESBL screening is the disk diffusion assay with clavulanic acid and cefotaxime and/or ceftazidime also called the double disc synergy test5 (Figure 1). ESBL producers are resistant to cefotaxime and ceftazidime. However, presence of clavulanic acid recovers the activity of cefotaxime and ceftazidime making the organism susceptible. If the activity of either one of these 3rd generation cephalosporin is recovered by clavulanic acid, then the presence of an ESBL is confirmed. A positive ESBL interpretation is resulted when there is ≥5 mm increase in zone diameter for either agent tested in combination with clavulanate versus the zone diameter of the agent tested alone. A broth microdilution approach is also possible. A positive ESBL interpretation would be a ≥3 ‘2-fold’ concentration decreases in an MIC for either agent tested in combination with clavulanate versus the MIC of the agent tested alone. Clinical Laboratory and Standards Institute (CLSI) guidance does not require ESBL testing for Enterobacterales but testing is recommended for infection prevention or specific institutional practices. For reporting of cephalosporin results, if current breakpoints are used for one or more cephalosporins, it is advised that the MICs are reported per usual. However, if obsolete cephalosporin breakpoints are used, then all penicillins, cephalosporins and aztreonam should be reported as resistant.6 It should be noted that there may be trivial differences between CLSI and European Committee on Antimicrobial Susceptibility Testing (EUCAST) guidelines and it is up to the individual institution to decide which guidelines to follow.
Table 1. Breakdown of the different generations of cephalosporins.
Other phenotypic based methods to detect for ESBLs include commercially available testing systems that have built in phenotypic ESBL screening. Performance varies depending on the manufacturer and ranges from 84-99% and 52-78% for sensitivity and specificity, respectively.7 Other commercially available assays include the colorimetric tests such as the Rapid ESBL Screen Kit (Rosco Diagnostica) that detects ESBL producers without differentiating between the various enzymes.5,8 This test has sensitivity of >90% when tested with cultured isolates or various specimens (blood, urine or respiratory) but variable specificity. There is also a lateral flow test (NG-Test CTX-M MULTI assay, NG Biotech, Guipry, France) developed to detect for CTX-M enzymes with reported sensitivity and specificity of >98%.9
Advancements in molecular testing have included ESBL genes as targets on commercially available diagnostic panels. As CTX-M is the most common and wide-spread ESBL gene, commercial, FDA-approved platforms including blood culture identification panels and pneumonia panels include the CTX-M marker as a target. Sensitivity ranging from 85-95% have been reported with variable specificity.10-12 Recently, the Acuitas AMR gene panel became the first FDA-cleared diagnostic test that includes a wide panel of 28 AMR markers including ESBL-related family of genes such as TEM and SHV with reported positive predictive agreement of 98.5% and 100%, respectively.13
Overall, there are various methods to detect for ESBL producers and detection for ESBL producers may not be a straightforward matter as there are many ESBL families of genes and in general, antibiotic resistance mechanisms in gram negative organisms are heterogeneous. However, given the rise in antimicrobial resistance, identification of ESBL producers is vital both in treatment of patients as well as surveillance.
REFERENCES
1. CDC. 2022. Special report – Covid-19 U.S. Impact on antimicrobioal resistance.
2. Castanheira M, Simner PJ, Bradford PA.2021. Extended-spectrum beta-lactamases: an update on their characteristics, epidemiology and detection. JAC Antimicrob Resist 3:dlab092.
4. Castanheira M, Simner PJ, Bradford PA.2021. Extended-spectrum β-lactamases: an update on their characteristics, epidemiology and detection. JAC Antimicrob Resist 3:dlab092.
5. Dortet L, Poirel L, Nordmann P.2015. Rapid detection of ESBL-producing Enterobacteriaceae in blood cultures. Emerg Infect Dis 21:504-7.
6. Fay D, Oldfather JE.1979. Standardization of direct susceptibility test for blood cultures. J Clin Microbiol 9:347-50.
7. Wiegand I, Geiss HK, Mack D, Stürenburg E, Seifert H.2007. Detection of extended-spectrum beta-lactamases among Enterobacteriaceae by use of semiautomated microbiology systems and manual detection procedures. J Clin Microbiol 45:1167-74.
8. Rood IGH, Li Q.2017. Review: Molecular detection of extended spectrum-beta-lactamase- and carbapenemase-producing Enterobacteriaceae in a clinical setting. Diagn Microbiol Infect Dis 89:245-250.
9. Bernabeu S, Ratnam KC, Boutal H, Gonzalez C, Vogel A, Devilliers K, Plaisance M, Oueslati S, Malhotra-Kumar S, Dortet L, Fortineau N, Simon S, Volland H, Naas T.2020. A Lateral Flow Immunoassay for the Rapid Identification of CTX-M-Producing Enterobacterales from Culture Plates and Positive Blood Cultures. Diagnostics (Basel) 10.
10. Murphy CN, Fowler R, Balada-Llasat JM, Carroll A, Stone H, Akerele O, Buchan B, Windham S, Hopp A, Ronen S, Relich RF, Buckner R, Warren DA, Humphries R, Campeau S, Huse H, Chandrasekaran S, Leber A, Everhart K, Harrington A, Kwong C, Bonwit A, Dien Bard J, Naccache S, Zimmerman C, Jones B, Rindlisbacher C, Buccambuso M, Clark A, Rogatcheva M, Graue C, Bourzac KM.2020. Multicenter Evaluation of the BioFire FilmArray Pneumonia/Pneumonia Plus Panel for Detection and Quantification of Agents of Lower Respiratory Tract Infection. J Clin Microbiol 58.
11. Peri AM, Ling W, Furuya-Kanamori L, Harris PNA, Paterson DL.2022. Performance of BioFire Blood Culture Identification 2 Panel (BCID2) for the detection of bloodstream pathogens and their associated resistance markers: a systematic review and meta-analysis of diagnostic test accuracy studies. BMC Infect Dis 22:794.
12. Klein M, Bacher J, Barth S, Atrzadeh F, Siebenhaller K, Ferreira I, Beisken S, Posch AE, Carroll KC, Wunderink RG, Qi C, Wu F, Hardy DJ, Patel R, Sims MD.2021. Multicenter Evaluation of the Unyvero Platform for Testing Bronchoalveolar Lavage Fluid. J Clin Microbiol 59.
13. Simner PJ, Musser KA, Mitchell K, Wise MG, Lewis S, Yee R, Bergman Y, Good CE, Abdelhamed AM, Li H, Laseman EM, Sahm D, Pitzer K, Quan J, Walker GT, Jacobs MR, Rhoads DD.2022. Multicenter Evaluation of the Acuitas AMR Gene Panel for Detection of an Extended Panel of Antimicrobial Resistance Genes among Bacterial Isolates. J Clin Microbiol 60:e0209821.
-Athulaprabha Murthi PhD is currently a Fellow at the NYC Public Health Laboratory. She is interested in antibiotic resistance and hopes to work towards better diagnostic testing and surveillance methods for monitoring resistance. She also enjoys writing and teaching whenever opportunity presents.
-Rebecca Yee, PhD, D(ABMM), M(ASCP)CM is the Chief of Microbiology, Director of Clinical Microbiology and Molecular Microbiology Laboratory at the George Washington University Hospital. Her interests include bacteriology, antimicrobial resistance, and development of infectious disease diagnostics.
In the United States, victims of gunshot wounds represent a significant majority of all homicides (and a high proportion of suicides). There’s a propensity among other medical specialties to think of forensic pathologists as “bullet pullers,” just collecting the used projectiles and moving on to the next case. However, autopsies of multiple gunshot wound victims can be some of the most detailed examinations we perform. Even though the cause of death isn’t a mystery, thorough observation and documentation is crucial for other questions which may arise – was the victim immediately incapacitated? Did the shooter reload during the assault? How close to the victim were the shots fired? To address these questions, it’s first helpful to understand the basics of gunshot wounds.
Gunshot wounds can be penetrating (entering the body without exiting) or perforating (entering and exiting the body). Full body x-rays are taken in all gunshot wounds to identify retained projectiles, all of which must be recovered as evidence. It’s important to not use metal tools (like forceps or scissors) when removing a projectile, which may scratch metal and interfere with ballistics comparison.
Under typical circumstances, distinguishing entrance from exit wounds is straightforward. Classic entrance wounds are circular, as the bullet hasn’t yet been deformed, and there is a surrounding rim of abrasion where the edges of the bullet scrape against the skin. In contrast, exit wounds have a stellate or slit-like appearance, but the wound edges can typically be reassembled and are not abraded. As with any area of medicine, though, real life doesn’t always follow the rules. An entrance wound can be atypical if the bullet has passed through an ‘intermediary target’ before striking the victim (say a piece of furniture or a car door). If the area of exit is pressed against a firm object (even a tight pants waistband), the skin edges will be abraded (or “shored”). Sometimes, the entrance and exit truly cannot be distinguished – this is more likely with superimposed decomposition, insect activity, or superficial wounds.
Range of fire is another important feature to note and is the reason we always take photographs of a wound before cleaning the body. When a gun is fired, smoke, soot, and unburnt particles of gunpowder exit the barrel as well as the bullet. The smoke creates “fouling,” dark discoloration which easily wipes away and can be seen if the end of the barrel is within approximately one foot of the victim. Stippling, caused by unburnt grains of gunpowder, are actual abrasions and can be seen when the projectile fired within 18” of the victim. These are gross generalizations, though, and in each individual circumstance the weapon itself must be tested to identify the distances. If soot or gunpowder particles aren’t visible on the skin surface, they may have been deposited on the victim’s clothing. ‘‘Bullet wipe’ is slightly different from fouling, in that it is dirt and residue on the actual bullet which gets ‘wiped’ off around the entrance wound on clothing – so it doesn’t tell you range of fire, but it can be helpful to identify tricky entrance wounds.
Now, for some of the common misconceptions around gunshot wounds …
Can you tell the caliber of the gun from the size of the wound? The answer is an emphatic “no.” Much of the injury caused by a bullet comes from the temporary wound cavity created by dissipation of kinetic energy; so no matter the size of the bullet, tissue will stretch and distort around it.
Can you tell the order in which gunshot wounds were sustained? Most often, no. In some autopsies, the first wounds cause so much blood loss that the later wounds lack hemorrhage, but this is the exception rather than the rule.
Can I tell what position the victim was in when they were shot? Again, usually not. In isolation, an autopsy can only tell you the trajectory of the bullet through the body; to determine the position of the victim when they were shot requires knowledge of either where the gun was when it was fired or where the bullet landed–two factors which are often not available.
These are just a few of the complexities faced by a pathologist when working with gunshot wounds, and we haven’t even covered different types of ammunition or firearms. Stay tuned for more in the future!
This is an example of a classic entrance gunshot wound – nearly circular, with a thin rim of abraded (or scraped) skin.In contrast, this exit wound is slit shaped, and the wound edges can be neatly reapproximated.This entrance wound has stippling on the surrounding skin; occasionally gunpowder particles are still visible embedded in the abrasions.
-Alison Krywanczyk, MD, FASCP, is currently a Deputy Medical Examiner at the Cuyahoga County Medical Examiner’s Office.
In television and movies, you’ll sometimes see a scene where the pathologist describes to a detective the specific dimensions of a knife used in an attack, all based on the autopsy findings. While this is no doubt extremely helpful to an investigation (and the limits of a 60-minute run time) it’s also impossible to provide this level of detail from an autopsy. Unfortunately the pervasiveness of such creative liberties in pop culture creates unrealistic expectations from juries, lawyers, and law enforcement, which we often need to gently correct. There are certain characteristics we can identify which may clarify what kind of blade was used, but conversely may not match the dimensions of the weapon at all.
While any object with a pointed or edged tip can be used to inflict sharp force injuries, we will focus on knives as the stereotypical ‘sharp force’ weapon. As a reminder, sharp force injuries have clean, neat edges and no tissue bridging in the wound depths. These are the features that are used to distinguish sharp force from lacerations, which are skin tears due to blunt force injury (see last month’s blog post for a refresher!). The most important first step for any instance of sharp force injury is to fully x-ray the body; this allows for identification of potential evidence to discover (such as a broken knife tip), and identifies potential sharp hazards for the prosector.
The first issue when interpreting sharp force injuries at autopsy is distortion of wounds by skin elasticity. Depending on the location and orientation of the wound, the edges may be pulled by tension along Langer’s lines. If this is the case, re- approximating the wound edges is needed before measuring for better accuracy. The ends of a stab wound may reflect whether a blade has one or two edges, depending on whether both ends are tapered (two-edged) or if one end is blunt and the other tapered (single-edged). The morphology isn’t always clear, though. Occasionally a double-edged blade will have a blunt-edged segment known as the “ricasso” just before the crossguard or handle. If the blade is fully inserted, the impression of the ricasso may make the wound appear to have two blunt edges.
The second difficulty faced is that knife wounds are rarely inflicted on a stationary body – there is typically (at least initially) a struggle involving motion of both the victim and the blade. This can result in wounds which are distorted from the dimensions of the weapon. These kinds of wounds also don’t necessarily imply that the person holding the knife was ‘torturing’ the victim – it just means that there was motion between the body and the blade. For example, a ‘dovetail’ or “V”-shaped wound can be created if the knife is inserted and removed at different angles – this could happen if either the victim is moving, if the assailant moves the angle of the knife, or both.
Another relatively frequent source of confusion is depth of wound relative to the length of the blade. All soft tissues are compressible, but certain parts of the body have a little more give. For example, the relatively soft abdominal pannus and viscera are easily compressed, whereas the chest is held in a rigid position by the rib cage. Therefore it’s very possible for a 5” blade to leave a wound 8” in depth if inflicted in a soft location like the abdomen. Additionally, if a blade isn’t inserted to the full length, the wound depth could also be shorter than the weapon.
One concerning trope on television is the immediate reconstruction of a crime based on autopsy findings. As one example, much emphasis is placed in the media on identifying ‘defensive wounds’. However, there is no way to tell at autopsy if a wound was sustained defensively or aggressively (and to be fair, even if the incident was caught on camera there could be differing interpretations). There are characteristics which are more commonly seen in ‘defensive’ wounds, such as locations on the hands and forearms as the victim attempts to block or grab the weapon. Hand wounds can also be seen on the assailant, though, as blood can make their hand slip onto the cutting blade during thrusts. Similarly, it is never possible to ascertain the “handedness” of an assailant from the wound pattern.
Serrated knives may leave a characteristic pattern of abrasions on the body but may leave no mark depending on the angle of the wound. So, if such marks are present, it’s distinctive of a serrated weapon – but if they’re absent, the weapon could be either a serrated or straight edged blade.
Contrary to what is shown on TV, there is no way to definitively ‘match’ one specific knife to a particular wound. The closest we could get would be finding a broken segment of the blade within the body – and even then, one must imagine there is more than one knife in the world that has a broken tip. While the world would be a much simpler place if such answers were possible, it’s our duty as scientists and physicians to be honest about the limitations of science and avoid speculation.
This is a stab wound inflicted by a knife with a single-edged blade, characterized by a squared-off, blunt end at the top of the photo and a tapered, “sharp” end at the bottom of the photo.This stab wound initially has very unclear morphology (on the left) due to tension placed on the wound by skin elasticity and nearby sutures. Once the wound is reapproximated (on the right), there are clearly two tapered edges which may be consistent with a double-edged blade.
-Alison Krywanczyk, MD, FASCP, is currently a Deputy Medical Examiner at the Cuyahoga County Medical Examiner’s Office.
With warmer weather approaching (or already arrived, depending on your location), it’s a good opportunity to review investigation of drowning deaths in forensic pathology. Drowning is the leading cause of deaths for children between the ages of 1 and 4 in the United States, but it can affect any age group.
Drowning is a diagnosis of exclusion – there are no pathognomonic signs of drowning, and a complete autopsy is required to rule out competing causes of death. Keeping an open mind during the investigation is the first step – discovering a body in water doesn’t automatically mean the cause of death is drowning. The body of a homicide victim may be disposed of in water as an attempt to destroy evidence, or someone may die of a cardiac arrythmia while they happen to be swimming.
The questions to answer in possible drowning deaths are much like those when faced with a body found after a fire (see “The Basics of Deaths by Fire” from February 23, 2023). Was the person alive when their body entered the water? And if they were, did they die from drowning – or did they die of another cause, and then become submerged?
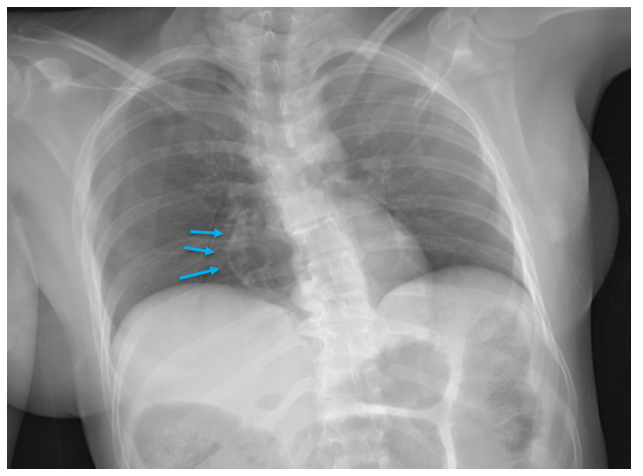
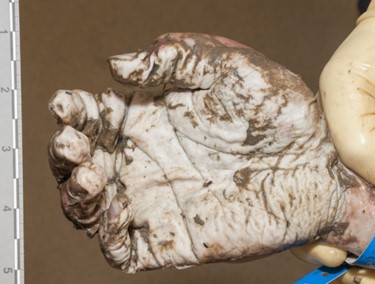
Autopsies of drowned individuals commonly reveal findings supportive of, but not specific for, drowning. The lungs are typically over-expanded and edematous, and foamy fluid may be in the airways. A “foam cone” may protrude from the nostrils and/or mouth. Pulmonary effusions can be present, and the petrous ridges at the base of the skull may show red-purple discoloration due to vascular congestion and hemorrhage. The stomach and sphenoid sinus may contain large amounts of watery fluid. Wrinkling and pallor of the skin of the hands and feet (formerly called “washerwoman’s hands”) is often identified but doesn’t necessarily indicate drowning as it can occur with pre-mortem or post-mortem submersion.
A posterior neck dissection is necessary in most drownings to rule out high cervical spine injuries, which are often overlooked without this special autopsy technique. This type of trauma can be seen in divers or jumpers who strike head-first in shallow bodies of water and may cause death by itself or contribute to the decedent’s inability to self-extricate from the water. Pathologists also need to be aware that post-mortem injuries can happen as the body is passively carried by currents and bumps into rocks or other debris, known as “travel abrasions”.
The body of water is another consideration. It would be highly unlikely for a neurologically alert teenager or adult to drown in a bathtub, whereas an infant could easily drown if left unsupervised. In contrast, it may take a river or ocean with strong currents to overpower experienced swimmers. Personal medical history is important, as well – an adult with epilepsy could drown even in shallow water if they experience a seizure. The temperature of the water can also play a role. Cold water can trigger cardiac arrhythmias, contribute to fatigue of skeletal muscles, or incite hypothermia leading to a loss of consciousness. Toxicology testing is an important ancillary test in drowning deaths to provide context and may reveal intoxications that help explain someone’s inability to remove themselves from the water. Alcohol can contribute to the impairment of physical coordination and/or increase risk-taking behaviors, and has been associated with up to 70% of water recreation-associated deaths.
If the autopsy doesn’t show any indicators of drowning but reveals potentially lethal natural disease (such as severe coronary artery stenosis), then it’s likely the person died while they happened to be in the water and not because they were in the water. In every situation, though, the autopsy findings must be correlated with the decedent’s history, the results of scene investigation, and toxicology testing before a final diagnosis can be rendered. In this way, autopsies of water-associated deaths highlight the importance of context and investigation in forensic pathology.
Figure 1. A classic example of the “foam cone” seen at autopsy in instances of drowning. This finding results from marked pulmonary edema, and isn’t specific for drowning – it can be seen in many other conditions including opiate overdoses and heart disease.Figure 2. Foamy fluid in the trachea and mainstem bronchi can be seen in any condition that causes pulmonary edema, and also is not specific for drowning.Figure3. Wrinkling and paleness of the hands and feet is often seen in bodies recovered from water, whether or not the cause of death was actually drowning.
References
Armstrong EJ, Erskine KL. Investigation of drowning deaths: a practical review. Academic Forensic Pathology, Jan 2018.
Against the backdrop of COVID-19, the world experienced a multicounty outbreak of Mpox (formally monkeypox) beginning in May of 2022. Prior to that time, the virus was primarily known to circulate within central and west African nations causing zoonotic disease. Clinical presentations of Mpox comprise signs and symptoms including rash on the hands, feet, face or mucous membranes and patients may experience fever or an influenza-like illness.1 Historically, transmission was associated with travel to an endemic region and contact with an infected animal. Importantly, the outbreak in 2022 was associated with broad changes in Mpox epidemiology, as most infections were acquired via sexual transmission.
Pox viruses and Mpox
Pox viruses are members of the family Poxviridae, which are double stranded DNA viruses that replicate entirely in the cytoplasm of host cells. They have worldwide distribution and cause disease in humans and other animals. Infection typically manifests as the formation of lesions, skin nodules or rash. Mpox belongs to the genus Orthopoxvirus which also includes other clinically important viruses including variola virus (smallpox), vaccinia virus, and cowpox. In the context of diagnosis, differentiation between the members of the Orthopox family becomes important.
The duration of illness with Mpox is usually between 2-4 weeks, with a variable incubation time most often between 6-13 days. The Mpox rash has historically been more focused on the face and extremities,2 and will cycle through stages including encrustation, scabbing, and eventually resolution. During the 2022 outbreak, an increasing number of presentations involved the anogenital and oral regions, further highlighting the change in epidemiology. The window for transmission is currently an area of active research as new data suggests transmission can begin prior to the appearance of symptomology.3
Diagnosis – Molecular
Mpox is generally diagnosed using PCR testing from a swabbed lesion. At the onset of this emerging infectious disease, the CDC shared its algorithm and testing for Mpox with public health laboratories. The first-generation algorithm largely reflected its potential use as a tool for screening for bioterrorism agents, which included using two-tiered testing. The first test was designed to demonstrate that Orthopox DNA was present and rule out variola virus by targeting the Orthopox DNA polymerase gene found not present in Variola (E9L-NVAR). The second step was to target an Mpox-specific gene encoding the envelope protein (B6R).4 It soon was readily apparent that the only Orthopox virus in circulation was Mpox, so the CDC updated its guidance in late June 2022 to confirming diagnosis of Mpox with the single Orthopox DNA-polymerase PCR assay.
However, despite this modification to improve expediency and like the situation faced at the onset of the COVID-19 pandemic, the need for testing greatly exceeded what public health infrastructure could support. Thus, laboratories designed and validated laboratory developed tests (LDTs) to expand access to testing, thus enabling physicians to interrogate the causes of a patient’s rash more thoroughly. This flexibility was essential given rising cases numbers and relatively non-specific symptomology of Mpox. By May 2023, over 80 laboratories registered Mpox LDTs with the Food and Drug Administration,5 and commercial device manufacturers are now including it in new and forthcoming assays still in development.
Diagnosis – Histopathology
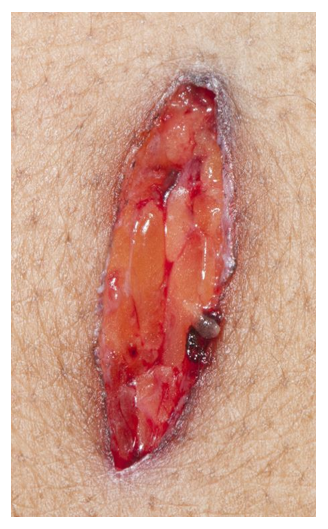
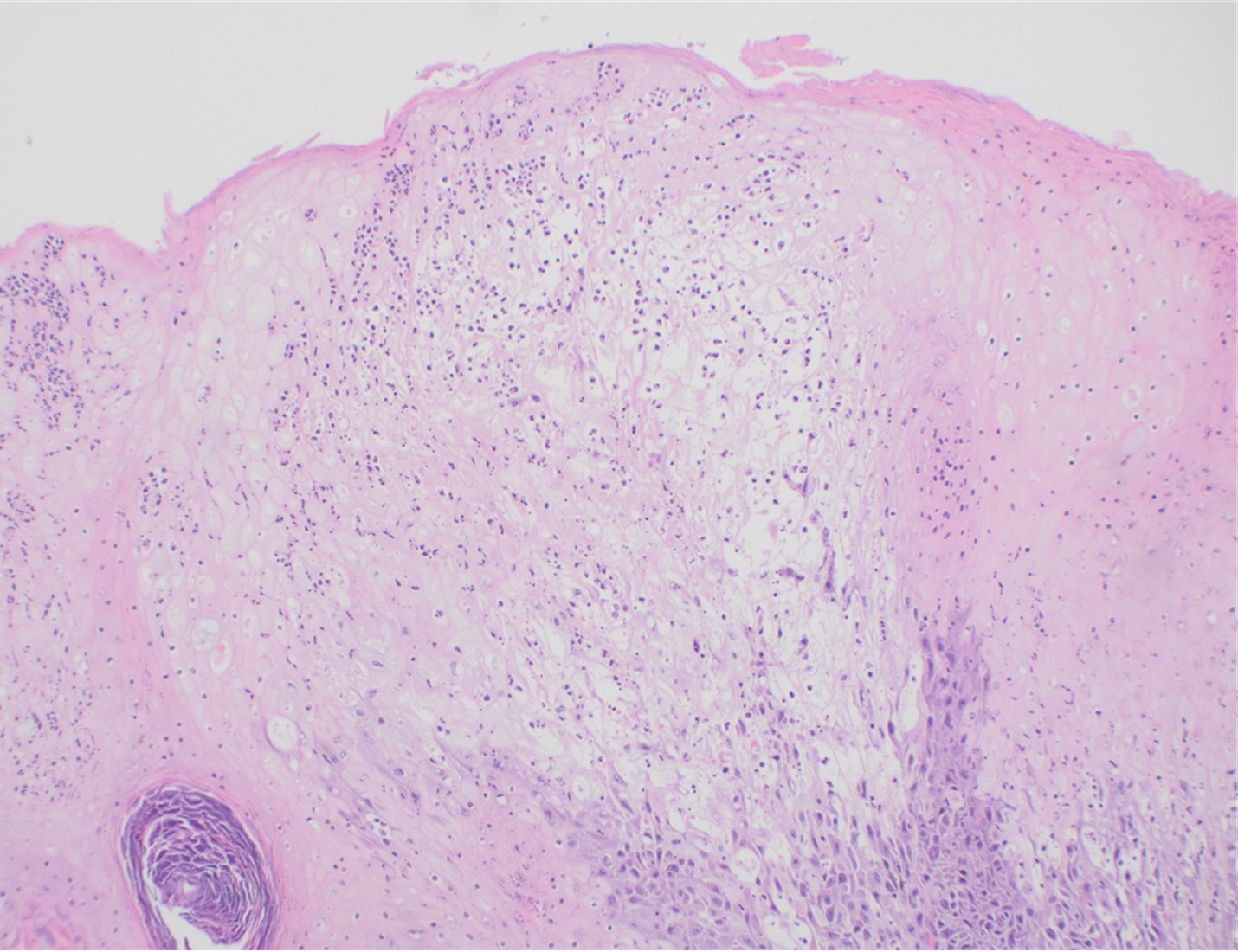
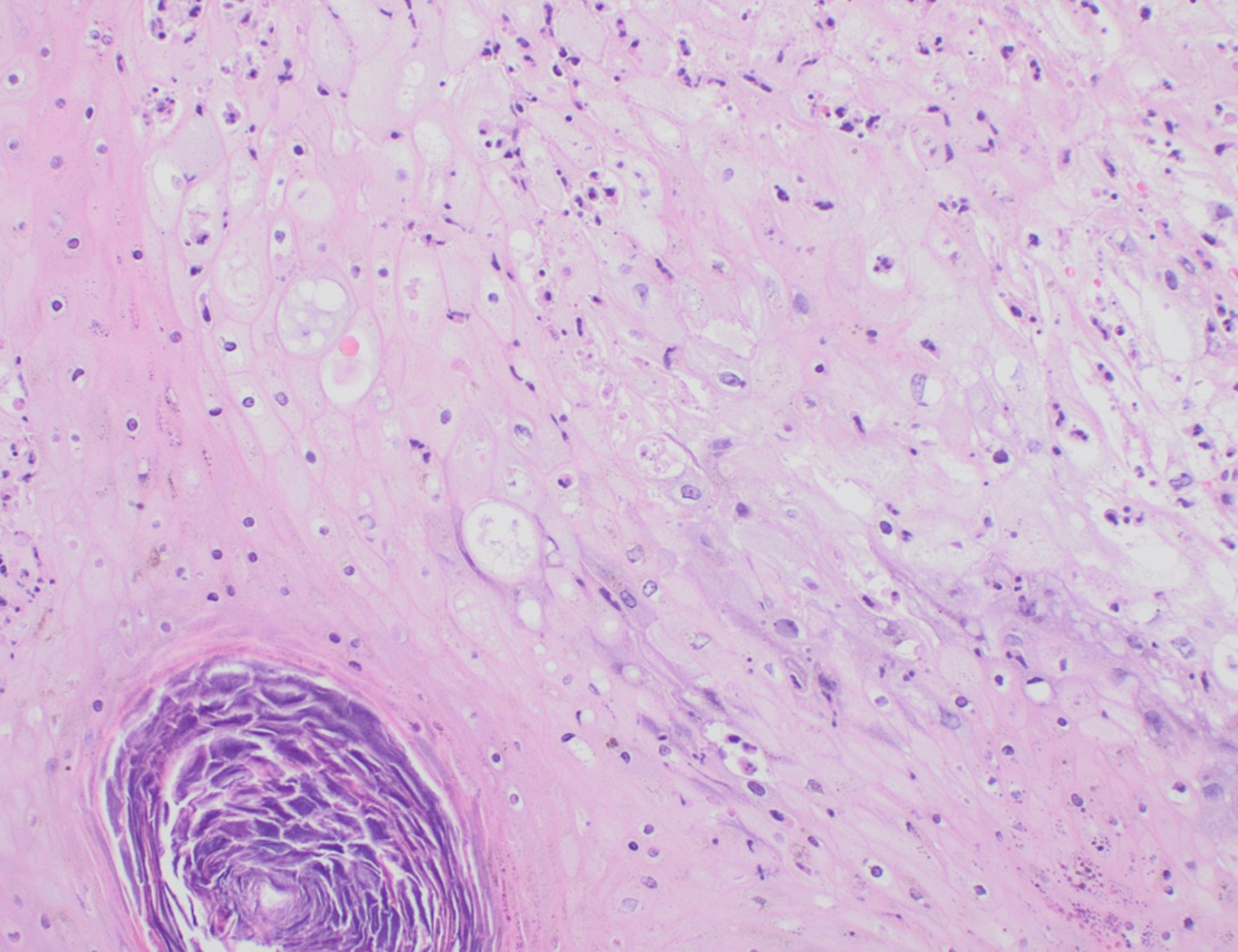
Although PCR testing is the mainstay of diagnosis, histopathologic evaluation of biopsy material from a lesion can also provide insight into the viral etiology. Mpox infected skin biopsies demonstrate similar histopathologic features of infections caused by other pox viruses. As the rash continues to evolve over time, representative histopathological changes can also be observed. Early lesions may demonstrate ballooning degeneration, acanthosis and spongiosis. More mature lesions progress to near total keratinocyte necrosis with exocytosis comprised of mixed cellular inflammatory infiltrate.6 Eosinophilic bodies may be identifiable in the cytoplasm of infected cells, commonly known as Guarnieri bodies, represent the mature virions produced in the cytoplasm of infected cells.
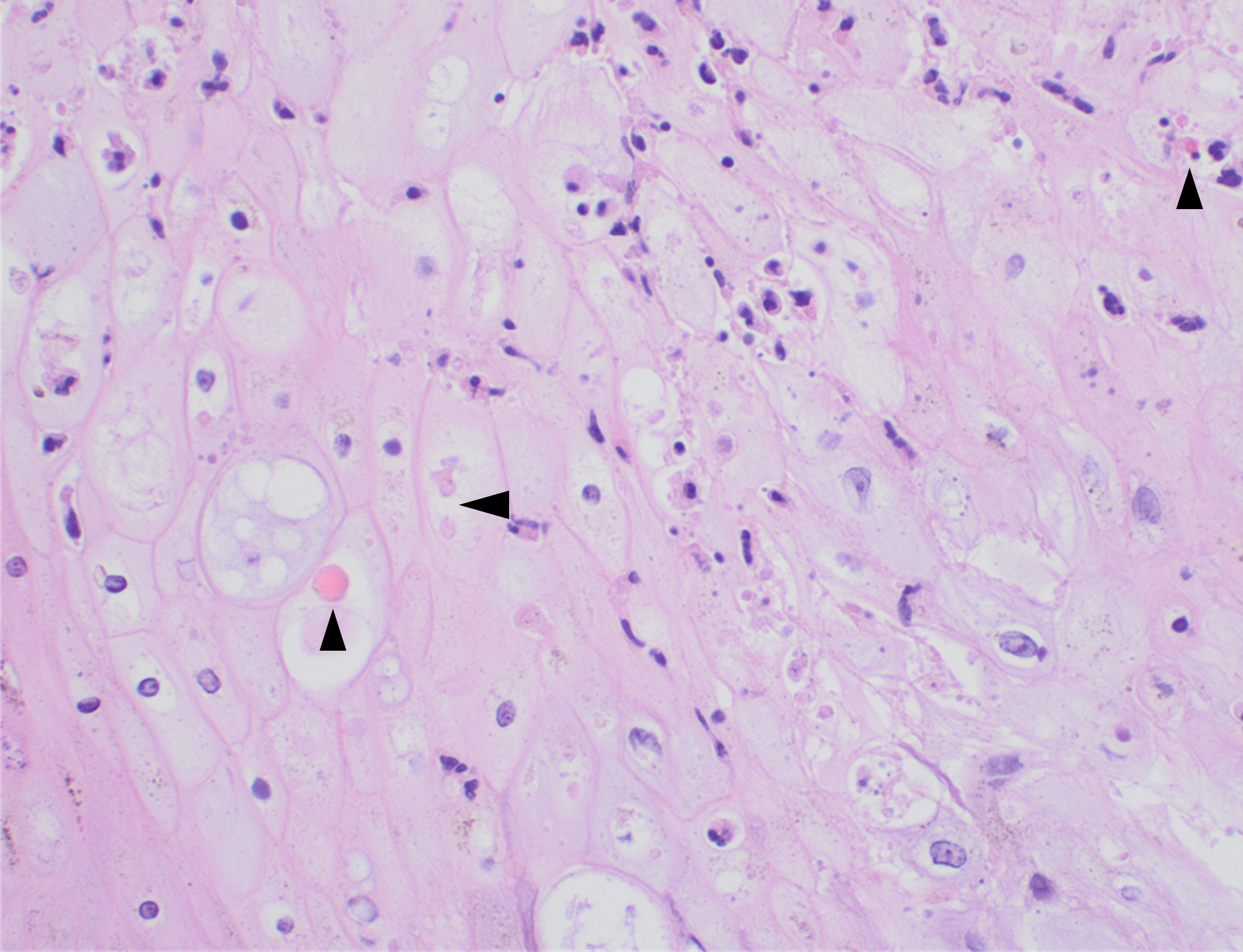
Recently, the histopathological description of 20 outbreak-associated clinical cases of Mpox from Spain was reported. Epidermal necrosis and keratinocytic ballooning were commonly encountered microscopic features associated with Mpox lesions.7 Figure 1 is a skin biopsy from a patient who presented with a vesicular eruption in September with a history of mpox, syphilis and herpes simplex infection whose lesions were worsening. It similarly shows ballooning degeneration, epidermal necrosis, exocytosis of neutrophils into the epidermis, and intracytoplasmic eosinophilic inclusions (Guarnieri bodies) (Figures 2-3).
Figure 1. Histopathology of MPOX from a biopsied skin lesion (4x magnification, H&E). Intact epidermis with evidence of ballooning keratinocyte degeneration and infiltration of neutrophils.Figure 2. Histopathology of Mpox (10x magnification, H&E). Epidermis with a cross-section of follicular infundibulum (hair follicle) is in the bottom left. The keratinocytes to the right demonstrate marked vacuolar change and small eosinophilic bodies can be observed in a background of neutrophils and necrotic keratinocytes.Figure 3. Histopathologic findings of MPOX in a biopsy of a skin lesion (40x magnification, H&E). High power magnification of viral inclusions, guarnieri bodies, (arrowheads) in a background of necrotic keratinocytes and neutrophilic infiltrate.
Treatment
Mpox is much milder than smallpox despite similar rash manifestations. In cases of severe Mpox infection, therapies used for smallpox have been compassionately utilized, but supportive measures are the mainstay of management of uncomplicated cases. Vaccination is now available as both a pre-exposure prophylaxis and post-exposure prophylaxis. It is important to note that the clinical effectiveness of the currently used vaccine in the United States is not known; however, early data across 32 US jurisdictions showed that among males 18-49, those who were unvaccinated had an Mpox incidence 14 times higher than similarly aged males who received at least one dose of vaccine at least 2 weeks prior.8
Conclusion
The Mpox outbreak, declared a global health emergency in July of 2022, has reinforced the need for flexibility within laboratories and industry to respond to emerging infectious diseases. The global health emergency for Mpox was declared over on May 11, 2023, but cases are still going to sporadically occur and minor outbreaks will result. The rapid development of numerous LDTs was essential to support the overwhelmed public health infrastructure, and this continued flexibility is needed to appropriately respond to future public health emergencies.
Rodríguez-Cuadrado FJ, Nájera L, Suárez D, Silvestre G, García-Fresnadillo D, Roustan G, Sánchez-Vázquez L, Jo M, Santonja C, Garrido-Ruiz MC, Vicente-Montaña AM, Rodríguez-Peralto JL, Requena L. Clinical, histopathologic, immunohistochemical, and electron microscopic findings in cutaneous monkeypox: A multicenter retrospective case series in Spain. J Am Acad Dermatol. 2023 Apr;88(4):856-863. doi: 10.1016/j.jaad.2022.12.027. Epub 2022 Dec 26. PMID: 36581043; PMCID: PMC9794029.
-Clare McCormick-Baw, MD, PhD is an Assistant Professor of Clinical Microbiology at UT Southwestern in Dallas, Texas. She has a passion for teaching about laboratory medicine in general and the best uses of the microbiology lab in particular.
-Travis Vandergriff, MD is an Associate Professor and Board-Certified Dermatopathologist and practicing Dermatologist at UT Southwestern Medical Center.
-Andrew Clark, PhD, D(ABMM) is an Assistant Professor at UT Southwestern Medical Center in the Department of Pathology, and Associate Director of the Clements University Hospital microbiology laboratory. He completed a CPEP-accredited postdoctoral fellowship in Medical and Public Health Microbiology at National Institutes of Health, and is interested in antimicrobial susceptibility and anaerobe pathophysiology.
I was recently reviewing new toxicology reports from my pending autopsies, and came across a report with the following results:
Looking at this in isolation, it would be easy to assume this person died from an overdose. Even low levels of fentanyl can be dangerous to an opioid-naive individual – a level this high is rare. Then there’s the added presence of fluoro fentanyl, a fentanyl analog, which would seem to support the notion of an overdose. The problem with this assumption? This person died from blunt force trauma as a pedestrian struck by a car. He was, according to witness accounts, walking and talking right until the moment of impact. Autopsy had shown multiple blunt force injuries incompatible with life.
This situation illustrates some of the complexity of postmortem forensic toxicology. Despite methodology being nearly the same, toxicology in a forensic setting differs in many important ways from that performed in a clinical setting.
The first major difference occurs in the pre-analytical phase. The results of clinical testing may be used to alter therapy or make a diagnosis. However, forensic toxicology results are meant to be used in a court of law, meaning the chain of custody needs to be maintained. If there is no documentation of who touched the sample and when, the integrity of the specimen can be called into question and results may be impermissible.
Not all forensic toxicology is performed on deceased patients. Specimens may be taken from the living during evaluation of an alleged assault, driving under the influence, or for workplace monitoring. In autopsy specimens though, postmortem redistribution (PMR) is another pre-analytical factor to consider. After death the stomach, intestines, and liver can serve as a drug reservoir and passively transfer the drug to surrounding vasculature. Other organs can also act as reservoirs, depending on where the drug is concentrated in life. Drugs which are highly lipid-soluble and/or have a high volume of distribution will diffuse down their gradient from adipose tissue into the bloodstream – antidepressants are notorious for this, and elevated postmortem levels need to be interpreted with caution.
Autopsy specimens are also more varied in type and quality than typical clinical specimens. Vitreous fluid, bile, and liver tissue are commonly collected at autopsy, in addition to central (heart) and peripheral (femoral or subclavian) blood. Femoral blood vessels, being relatively isolated from PMR-causing drug reservoirs, are a preferred source of specimens. Decomposition or trauma can limit the types or quantity of specimens and may even alter results. After death, bacteria from the GI tract proliferate and can produce measurable levels of ethanol in the blood. Decomposition also produces beta-phenethylamine, which can trigger a ‘positive’ result for methamphetamine on ELISA-based tests.
The post-analytical phase of autopsy toxicology also poses unique challenges. Lawyers and law enforcement will sometimes ask what the ‘lethal level’ of a drug is, and they’re invariably disappointed by my response. While there are published ranges of toxicity and lethality for most drugs, these are only general guidelines. There is no absolute lethal blood level for prescription or illicit drugs. Opioid users develop tolerance, making them relatively immune to a dose which would kill an opioid-naive person. In the example of the pedestrian described above, he had a long history of heroin abuse and could therefore tolerate much higher levels than most. For stimulants like cocaine and methamphetamine, there are no documented ‘safe’ levels as any amount could act as an arrhythmic agent. To add to the complexity, most overdose deaths involve multiple substances which may have synergistic effects and interactions that are difficult to parse.
Because of the reasons given above, the National Association of Medical Examiners still recommends full autopsy for possible overdoses. Deciding if a death was due to overdose is more complex than just reading a toxicology report – it requires interpretation and correlation with the autopsy findings and overall investigation.
References
D’Anna T, et al. The chain of custody in the era of modern forensics: from the classic procedures for gathering evidence for the new challenges related to digital data. Healthcare. 2023 Mar;11(5):634.
Davis GG, et al. National Association of Medical Examiners Position Paper: Recommendations for the Investigation, Diagnosis, and Certification of Deaths Related to Opioid Drugs. Acad Forensic Pathol 2013 3(1):77-81.
Pelissier-Alicot AL, et al. Mechanisms underlying postmortem redistribution of drugs: A review. J Anal Toxicol. 2003 Nov-Dec;27(8):533-44.