Case History
The patient is a 2.5 year old male who is being evaluated for a liver transplant versus biliary diversion surgery. The patient was born at 2 kilograms and went home with mom one week after birth. The patient was readmitted back to the hospital for evaluation of jaundice and since then the patient has been intermittently hospitalized for episodes of worsening jaundice, acholic stools, scleral icterus, and pruritus. At 5 months of age, the patient was diagnosed with progressive familial intrahepatic cholestasis, type 2, and was placed on the liver transplant list. As a result of the liver failure, the patient has developed coagulopathy, hypocalcemia resulting in seizures, and pruritus. The family history is significant for no known congenital liver diseases.
The father was worked up for living donation and was found to be a suitable donor, and is donating the left lateral segment of his liver.
Diagnosis
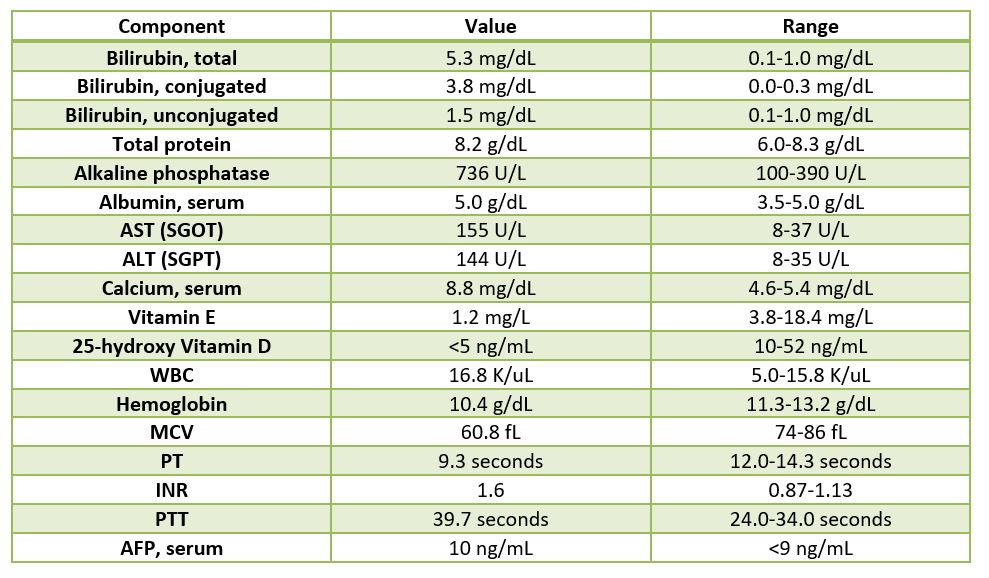
Received in the Surgical Pathology laboratory is a 700 gm, 23.5 x 14.5 x 3.5 cm explanted liver with an attached 4.5 x 1.2 x 0.4 cm gallbladder. The liver specimen has a smooth, green-red liver capsule without any grossly identifiable nodules or lesions (Image 1). The gallbladder has a yellow-pink external surface and is opened to reveal a 1.5 x 0.7 x 0.4 cm dark brown stone with a small amount of brown-yellow bile fluid. The liver is sectioned to reveal a smooth green-red cut surface (Image 2). No lesions are identified and minimal hilar structures are included with the specimen. Portions of the specimen have been taken for electron microscopy and frozen for future diagnostic purposes. Submitted sections include:
Cassette 1 and 2: Hilar structures
Cassettes 3-15: Representative sections of liver parenchyma
Cassette 16: representative section of gallbladder
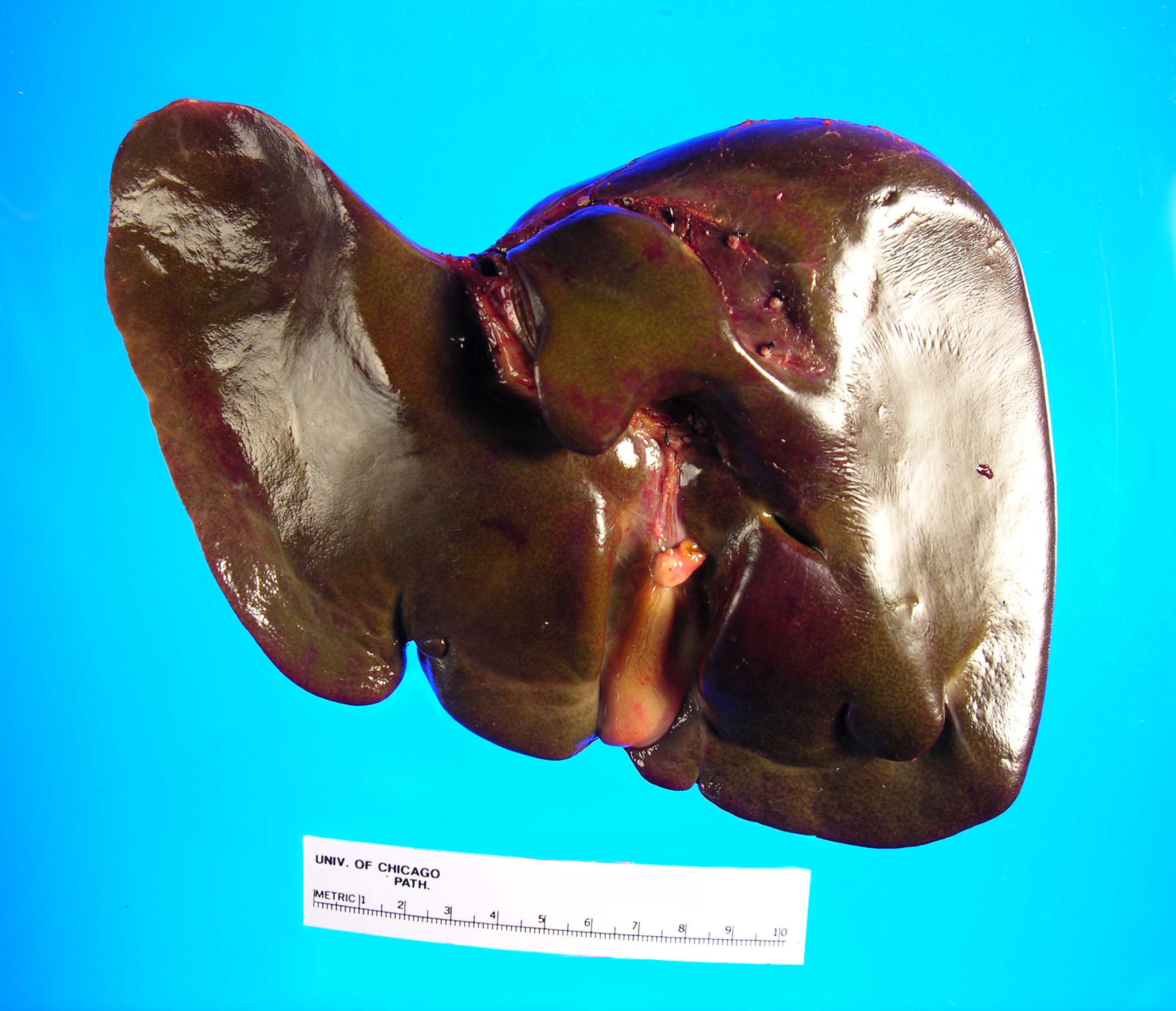

On microscopy, the trichrome stain highlights the presence of portal and centrilobular fibrosis, with focal bridging. However, regenerative nodule formation is not evident. The portal tracts contain sparse mononuclear cell infiltrates. Significant bile ductular proliferation is also evident, as confirmed by a CK7 immunostain. However, the native bile ducts appear unremarkable. There is also considerable hepatocellular and canalicular cholestasis in the centrilobular regions. Occasional multinucleated hepatocytes are also seen within the centrolobular zones. No steatosis is evident.
This constellation of histologic features is consistent with the clinical history of progressive familial intrahepatic cholestasis, type II.
Discussion
Progressive familial intrahepatic cholestasis (PFIC) is a group of autosomal recessive disorders that affects bile formation and results in cholestasis of the liver, usually beginning in infancy and childhood. There are three types of PFIC, each related to a mutation in the liver transport system genes that are involved in bile formation. PFIC type 1 (PFIC1), which is also referred to as Byler disease, is due to impaired bile salt secretion related to a ATP8B1 gene that encodes the FIC1 protein. PFIC type 2 (PFIC2), which is referred to as Byler syndrome, is due to impaired bile salt secretion (similar to type 1), but is related to the ABCB11 gene that encodes the bile salt export pump, or BSEP. PFIC type 3 (PFIC3) is due to impaired biliary phospholipid secretion that is related to a defect in the ABCB4 gene that encodes the multi-drug resistant 3 protein, or MDR3.
PFIC is suspected to be the cause of cholestasis in 10-15% of children, and is also the underlying cause of liver transplants in 10-15% of children. The exact prevalence remains unknown, but is estimated to be between 1 in every 50,000-100,000 births. PFIC1 and PFIC2 account for 2/3 of all PFIC cases, with PFIC3 making up the other 1/3. PFIC is present worldwide, and there does not appear to be a gender predilection.
The main clinical manifestation in all forms of PFIC, hence the name, is cholestasis, and will usually appear in the first few months of life with PFIC1 and PFIC2. Recurring episodes of jaundice are also present in PFIC1, whereas permanent jaundice and a rapid evolution to liver failure are characteristic of PFIC2. In PFIC3, cholestasis is noted within the first year of life in 1/3 of all cases, but rarely will be present in the neonatal period. PFIC3 can also present later in infancy, childhood or even early adulthood, with gastrointestinal bleeding due to portal hypertension and cirrhosis being the main symptoms that the patient would present with. Pruritus is severe in PFIC 1 and 2, but has a more mild presentation in PFIC3. There have been multiple cases reported of hepatocellular carcinoma that are associated with PFIC2, but there so far have not been any cases of hepatocellular carcinoma reported that are associated with PFIC3. Other signs and symptoms that may be present in PFIC1 include short stature, deafness, diarrhea, pancreatitis and liver steatosis. When examining clinical laboratory results, patients with PFIC1 and PFIC 2 will have normal serum gamma-glutamyltransferase (GGT) levels, but patients with PFIC3 will have elevated GGT levels. PFIC1 and PFIC2 can be differentiated from each other by the higher transaminase and alpha-fetoprotein levels that are found in PFIC2. When analyzing the biliary bile salt concentrations, PFIC1 will have mildly decreased levels (3-8 mM), PFIC2 will have drastically decreased levels (<1 mM), and PFIC3 will have normal levels. In addition, the biliary bile salt:phospholipid ratio and the cholesterol:phospholipid ratio will be approximately 5 times higher in PFIC3 than in normal bile, due to the biliary phospholipid levels being dramatically decreased (normal phospholipid range = 19-24%, PFIC phospholipid range = 1-15%).
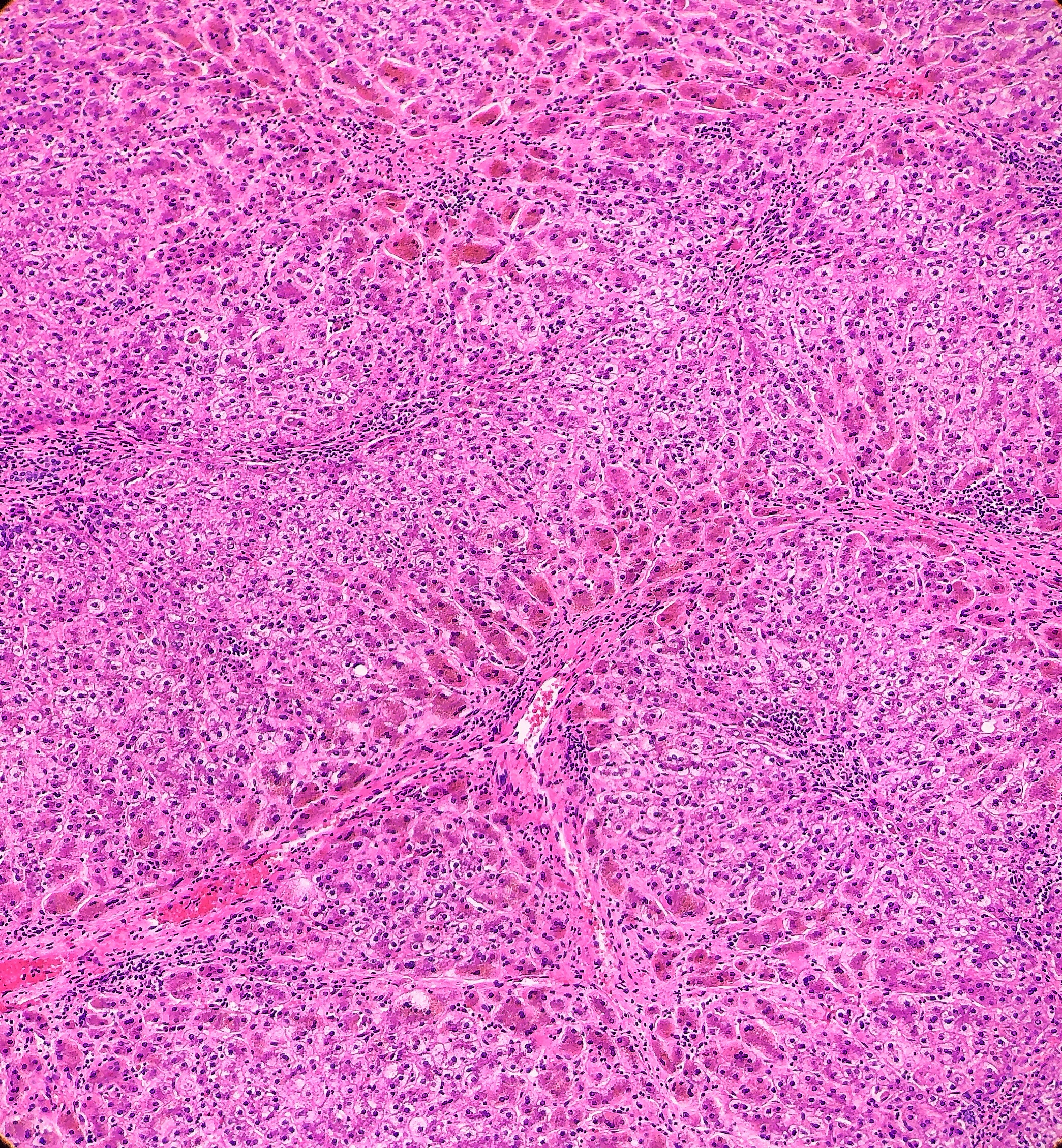
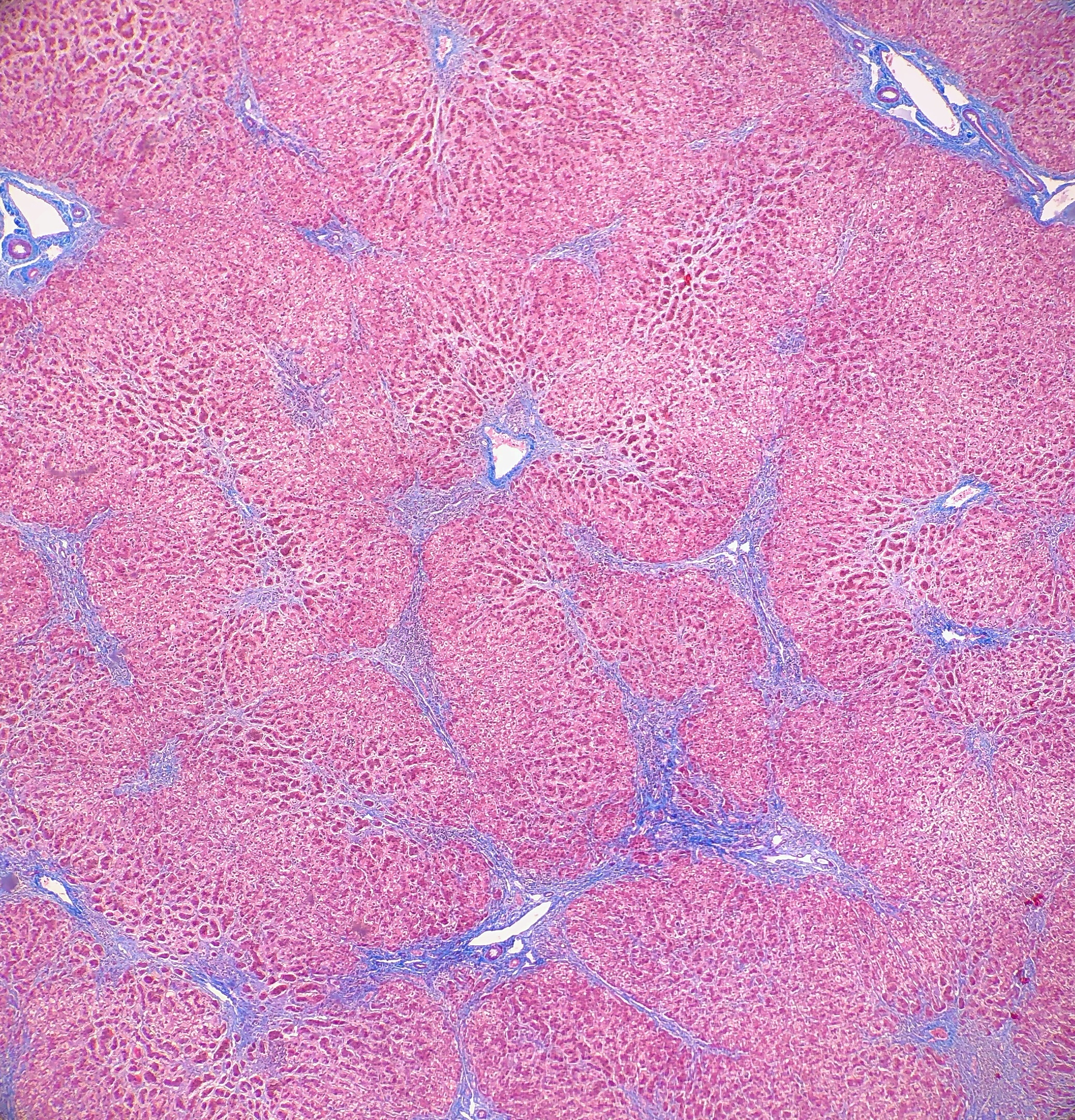
Histologically, PFIC1 and PFIC 2 will have canalicular cholestasis, an absence of true ductular proliferation, and periportal biliary metaplasia of the hepatocytes. In PFIC2, these manifestations are much more worrisome with more marked lobular and portal fibrosis, and inflammation, as well as having much more pronounced necrosis and giant cell transformation (Images 3 and 4). PFIC3 will show portal fibrosis and true ductal proliferation, with a mixed inflammatory infiltrate. In addition, cholestasis can be present in the lobule and in some of the ductules that contain bile plugs. Cytokeratin staining can help confirm the ductular proliferation within the portal tract. Mild or absent canalicular staining with BSEP and MDR3 antibodies will help to diagnose PFIC2 and PFIC3, respectively.
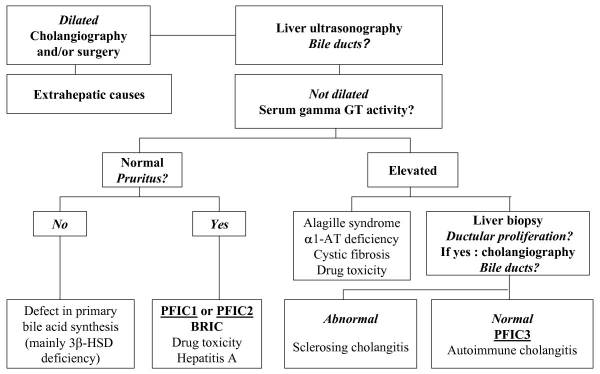
A diagnosis of PFIC is based on the clinical manifestations, liver ultrasonography, cholangiography and liver histology, as well as on specific tests for excluding other causes of childhood cholestasis (such as biliary atresia, Alagille syndrome, cystic fibrosis and alpha-1 antitrypsine deficiency). Ultrasonography of the liver will be normal with the exception of a possible dilated gallbladder. At the time of the liver biopsy, a portion of tissue can be submitted for electron microscopy, which in the case of PFIC, can show canalicular dilatation, microvilli loss, abnormal mitochondrial internal structures, and varying intra-canalicular accumulations of bile. PFIC1 will have coarsely, granular bile on electron microscopy, whereas PFIC2 will have a more amorphous appearance. If biliary obstruction is noted on the liver biopsy, a cholangiography will need to be performed to exclude sclerosing cholangitis. If a normal biliary tree is observed, as in PFIC, bile can be collected for biliary bile salt analysis (which was discussed earlier in the laboratory results section). Differentiating between PFIC1, PFIC2 and PFIC3 can be quite troublesome, but luckily Davit-Spraul, Gonzales, Baussan and Jacquemin proposed a fantastic schematic for the clinical diagnosis of PFIC, which is presented as Figure 1.
Ursodeoxycholic acid (UDCA) therapy should be considered in all patients with PFIC to prevent liver damage and provide relief from pruritus. Rifampicin and Cholestyramine can help in cases of PFIC3, but have been found to provide no improvement in PFIC1 or PFIC2. In some PFIC1 or PFIC2 patients, biliary diversion can also relieve pruritus and slow disease progression. The total caloric intake should be around 125% of the recommended daily allowance. Dietary fats should come in the form of medium chain triglycerides, and care should be taken to check the patient’s vitamin levels to look for signs of vitamin deficiency. Patients with PFIC2 should be monitored for hepatocellular carcinoma, beginning from the first year of life. Ultimately, most PFIC patients develop fibrosis and end-stage liver disease before adulthood, and are candidates for liver transplantation. Diarrhea, steatosis and short stature may not improve after liver transplantation, and could become aggravated from the procedure. Hepatocyte transplantation, gene therapy or specific targeted pharmacotherapy are possible alternative therapies for PFIC, but will require more research and studies to determine whether they are viable options.
References
- Davit-Spraul A, Gonzales E, Baussan C, Jacquemin E. Progressive familial intrahepatic cholestasis. Orphanet J Rare Dis. 2009;4(1). doi:10.1186/1750-1172-4-1
- Evason K, Bove KE, Finegold MJ, et al. Morphologic findings in progressive familial intrahepatic cholestasis 2 (PFIC2): correlation with genetic and immunohistochemical studies. Am J Surg Pathol. 2011;35(5):687–696. doi:10.1097/PAS.0b013e318212ec87
- Srivastava A. Progressive Familial Intrahepatic Cholestasis. J Clin Exp Hepatol. 2013;4(1):25-36. doi: 10.1016/j.jceh.2013.10.005

-Cory Nash is a board certified Pathologists’ Assistant, specializing in surgical and gross pathology. He currently works as a Pathologists’ Assistant at the University of Chicago Medical Center. His job involves the macroscopic examination, dissection and tissue submission of surgical specimens, ranging from biopsies to multi-organ resections. Cory has a special interest in head and neck pathology, as well as bone and soft tissue pathology. Cory can be followed on twitter at @iplaywithorgans.
