Hello everyone and welcome back!
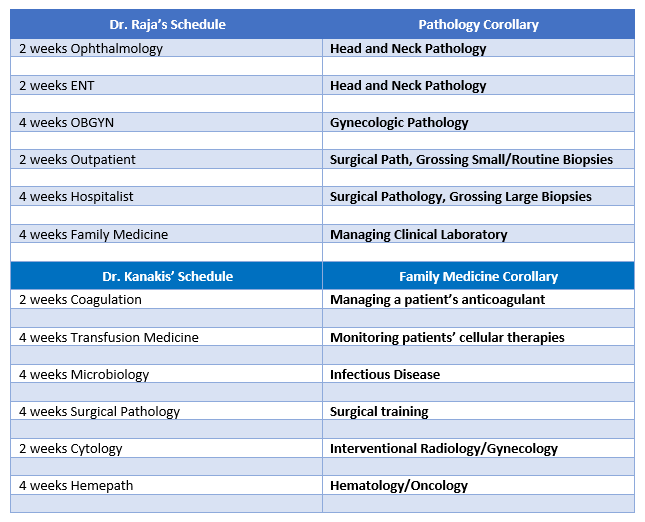
So here I am: a med school matriculant, top-choice program matched, doing exactly the training and work I’ve wanted to since before medical school. Totally made it! But before I go all “Fresh Prince of Maywood” on you, there’s a lot to unpack within the word “residency.” I’ve discussed this before, comparing pathology subspecialties with my primary care friend Dr. Raja’s rotation schedule, but you probably already know a little about what pathology residents do. I want to talk about what most folks might not know, why most residents absolutely disappear from their families or loved ones, and why going in with already greying hair isn’t going to make this any better, ha-ha…

I’m a resident at Loyola Medicine in Maywood, IL; I also went to undergrad (over a decade ago) at Loyola Chicago. So, for me, this is a very cool full-circle experience. I bring this symmetry up because a lot of people talk about the “culture” of an institution when they’re looking for a perfect match. I’ve talked about my experiences with hospitals’ institutional cultures before, at Bronx Care, Staten Island, Mayo Clinic, Danbury, Brooklyn, and more before medical school. I choose to highlight Loyola for you both because it’s already home to me, and also because it has a unique disposition. When you walk across the stage at graduation, Loyolans are instructed to “Set the world on fire”—a quote from the university’s namesake and patron St. Ignacious Loyola. It follows a Jesuit tradition of valuing education as one of the most powerful tools to address social inequality, injustice, poverty, and whatever ails our society. By being bold and passionate (like a fire, get it…?) true leadership can manifest in graduates’ futures.

But all graduations are decorated with pomp and circumstance. As graduates sit and wait for their name, they are pontificated at about the importance, poignancy, and grand scale of opportunity that awaits them. But what happens after graduating, college, or graduate school, or medical school? The answer varies widely for many, but I can speak to those who end up with the long white coats. I’ll be honest, allegorically, college is a lesson in walking, graduate school is a lesson in running, medical school is a lesson in cartwheels—after you’ve somewhat mastered this, the world that awaits you demands powerful cartwheels (with tricks) up multiple Mt. Everests, and you might be able to use the bathroom…you might. Haha, a little hyperbole. I mean I am SO glad pathology training isn’t like some other specialties (looking at you surgery…) but the demand is there, nonetheless. I would say ours might be more cerebral because, what we trade in for not having an intern year, we are “gifted” with having to lean 4+ years of material presented as an iceberg tip in medical school.
In a recent Inside the Lab podcast, the topic of burnout was discussed. (Check it out here!) Labratorians—and healthcare staff in every role—have been feeling the COVID push all year. More is expected of us, more is demanded of our system and its output, and there is no relief or break in sight. That prolonged demand on our expertise (and time) puts a significant strain on all our collective psyche’s. Nowhere is that more apparent than in healthcare. Paramedics run long uninterrupted shifts seeing tragic emergency one after another. Nurses do 12hr shifts back to back for days, especially when there isn’t enough staff to support days off (while patient census climbs higher and higher). But in medicine, poor medical school post-graduates are expected to literally “reside” in the hospital, ergo resident. The term came from the training model coined at Johns Hopkins in the early 1900s. And, up until a few years ago, the powers that be decided that residents should log no more than a maximum of 80 hours a week with the longest shift you can work 24 hours. Fun fact: the IRS, yes those guys, defines full-time work as 30-40 hours per week or 130 hours per month. If you work a “full time” job, you probably work 40 hours a week/160 hours a month. So, for young resident physicians: that two full time jobs, coming in hot at just about the average US salary of 55-60k. Outstanding. However, while I find myself lucky and would anecdotally say that I don’t think I’ll be getting any notifications or flags on logging too many hours at the hospital, the reality is that many physician trainees work right up to the maximum (and more). The old guard cites that 80 hours isn’t enough time to train a functioning physician, as they leave patient care at a sensitive time where they effectively abandon their learning. But …burnout. The “reduction” to 80 hours one would think reduces stress and burnout, but lo and behold a paper (from FIFTEEN years ago) says nu-uh. “Changes in parameters of resident and faculty emotional exhaustion, depersonalization, and personal accomplishment did not show statistical significance…Despite successful reductions in resident work hours, measures of burnout were not significantly affected.” (JAMA, 2004)

Regardless, those of us in postgraduate medical training are here for a reason. I identify as one of the few who finds himself in a lucky spot, where my institution—and my profession of choice—don’t demand that kind of hourly expectation of me. But many of my other colleges aren’t as lucky. Surgeons, internists, family doctors, and more are working themselves to the limit. And that doesn’t include anything about the COVID pandemic. Whether you’re a graduate of Loyola or not, we’re all expected to “set the world on fire,” I just hope we don’t burn out in the process. Stay in tune with your needs and your support system, learn to recognize signs of burnout as much more than fatigue, and remember to extend compassion to everyone—you never know what load they might be carrying. Remember those things and you can navigate a packed work-week…or a pandemic!
Thanks for reading!
See you next time!

–Constantine E. Kanakis MD, MSc, MLS(ASCP)CM is a first-year resident physician in the Pathology and Laboratory Medicine Department at Loyola University Medical Center in Chicago with interests in hematopathology, transfusion medicine, bioethics, public health, and graphic medicine. He is a certified CAP inspector, holds an ASCP LMU certificate, and xxx. He was named on the 2017 ASCP Forty Under 40 list, The Pathologist magazine’s 2020 Power List and serves on ASCP’s Commission for Continuing Professional Development, Social Media Committee, and Patient Champions Advisory Board. He was featured in several online forums during the peak of the COVID pandemic discussing laboratory-related testing considerations, delivered a TEDx talk called “Unrecognizable Medicine,” and sits on
the Auxiliary Board of the American Red Cross in Illinois. Dr. Kanakis is active on social media; follow him at @CEKanakisMD.