Pathology is a perfectly blended specialty filled with food, fun and a whole range of factual morphological descriptions!
As a pathology resident, one of the first things that got me intrigued by the specialty was its strong association with many food epithets. From the almond-shaped ovary,1 to the blueberry muffin baby,2 to the coffee bean nuclei in the thyroid,3 fried egg appearance of mast cells,4 grape-like lesions seen in molar pregnancy5 to the flat cake placenta6 and even to the strawberry cervix!7 The list is endless. I found these descriptions so interesting that I kept asking myself, “why do pathologists have to make associations with food for many normal and pathological disease processes we see around?”
Aside from the fun association with food (which I happen to like a lot), getting to learn and understand the pathology of disease processes, genetic and syndromic associations have been a fascinating, humbling, and altogether nerve-wracking experience for me.
It has been fascinating because I totally enjoy learning about the underlying processes that get some people sick while others stay healthy. At the same time, it has also been humbling, because, then I realize that so many disease processes are genetically determined and so out of our control. Along the same lines, the experience has also been neck-wracking, because of the detail and efficiency that goes into mastering different disease morphologies and preparing a comprehensive pathology report. The ability to tell the difference between two very similar disease entities but with different morphological features can drive one crazy, because, sometimes everything just seems to look the same!
I remember my early days as a resident. The first week in residency training to be precise. Then, I got reintroduced to the microscope, which is the power of the pathologist. Looking into the microscope and feigning to see what the senior residents and attendings were seeing felt like outright torture to me. You know why? It’s because everything under the microscope was either blue or pink.
In my few years of training as a resident, I have come to learn that in order to be successful as a pathologist, one must be adept with every single detail. As Pathologists, we deal with the facts. We do not make things up, and strive to present the facts of every case which ultimately supports our rendered diagnoses.
Unlike when I first started my residency training, I now know that not everything under the microscope is just blue and pink, and even if they are indeed blue and pink, the degree of their “blueness” or “pinkness” varies. And the intensity of the hematoxylin and eosin (H&E)/immunohistochemical stains may sometimes tell disease entities apart from one another. So, sometimes when people ask me what type of doctor I am training to be, I tell them, “I am a doctor of colors,” which of course often leaves them confused!
I also tell people that I am training to be a doctor who works from behind the scenes, to make sure they get treated right all the time. And this realization I believe is what has created the greatest impression for me. Realizing that a patient’s choice of treatment may totally be dependent on the pronouncements I make on their disease process, is something that gets me motivated to keep putting in my best into my training in order to become one of the best in my field. Therefore, even though we operate as doctors from behind the scenes, our professional judgments often go a long way in impacting the welfare and outcomes of patients whom we never get to see, which is one of the aspects of the specialty that I truly love.
So, pathology as a specialty has given me a more robust meaning to life. I have learned to value and appreciate the time I spend with those I love, and to make special moments with them count. It has made me realize that there are certain things about life such as genetic diseases, that I have no control over and therefore should only be concerned with giving my very best all the time. Pathology has also made me more detail oriented, by learning to distinguish benign from malignant processes. It has reinforced for me, the importance of being the best person I can be to both my family, neighbors and my community in general. And I would also add that pathology has further reignited my love for good food. So, let the party begin!!!
Mehta V, Balachandran C, Lonikar V. Blueberry muffin baby: a pictoral differential diagnosis. Dermatol Online J. 2008;14(2):8.
Oertli D, Udelsman R. Surgery of the thyroid and parathyroid Glands. Springer Science & Business Media; 2012. p. 620.
Bolognia JL, Jorizzo JL, Rapini RP. Dermatology. Gulf Professional Publishing; p. 1438.
Daftary. 100+Clinical Cases In Obstetrics. Elsevier India; 2006. p. 478.
Power ML, Schulkin J. The Evolution of the Human Placenta. JHU Press; 2012. p. 278.
Swygard H, Seña AC, Hobbs MM, Cohen MS. Trichomoniasis: clinical manifestations, diagnosis and management. Sex Transm Infect. 2004 Apr 1;80(2):91–5.
-Evi Abada, MD, MS is a Resident Physician in anatomic and clinical pathology at the Wayne State University School of Medicine/Detroit Medical Center in Michigan. She earned her Masters of Science in International Health Policy and Management from Brandeis University in Massachusetts, and is a global health advocate. Dr. Abada has been appointed to serve on the ASCP’s Resident’s Council and was named one of ASCP’S 40 under Forty honorees for the year 2020. You can follow her on twitter @EviAbadaMD.
I walked into the autopsy suite, trembling and drenched in sweat, even though the atmospheric temperature was as cool as it could ever be. It was my second autopsy experience as a pathology resident and I could not make out exactly how I was feeling. My first session had exposed me to the critical role of pathologists in solving complex clinical puzzles and had left me shaken for days. And, I still wasn’t sure how the second session was going to be. But, one thing I was sure of was the fact that I still felt uncomfortable.
Not uncomfortable because of the task that had been given to us to find out the cause of death of the person I was going to meet. But I felt very uneasy with the fact that I did not know what to expect, yet again. The first session had been that of a middle-aged woman. This was going to be a case of a young man. Two different scenarios and diagnoses. I did not know what to expect. My stomach turned and churned and I could also feel my heart thumping loudly in my chest.
I looked up at my senior resident, with my attending physician observing our every move. He looked very comfortable with what we were about to do. He seemed to approach the entire situation like it was a routine procedure for him. I questioned myself, “would I ever get comfortable with doing autopsies like him?”
I listened attentively as the senior resident walked me through the process of performing an autopsy and what our duties as pathologists was supposed to be. I tried to listen as my senior colleague who was obviously very familiar with the process gave me a detailed lecture. I felt my mind wandering away, even though it seemed as though I was paying attention to what he was saying. My attention drifted back and forth as I couldn’t help thinking about so many other things including the complexities surrounding life and death.
As we went through the organs and finally began working on the lungs and heart were his primary pathologies were supposed to be, I was amazed at the pathology I was being exposed to. His bilateral lungs were severely fibrotic, encased with numerous calcified nodules that eventually turned out to be non-caseating granulomas. He also had calcified hilar nodules also confirmed histopathologically as non-caseating granulomas and his heart was markedly enlarged, with hypertrophy of biventricular walls, more prominent on the right side. His pulmonary arteries also showed signs of severe vascular disease with hyalinization and fibrosis. He had disseminated sarcoidosis, with his heart and lungs more severely affected. The sarcoid granulomas had spared the other organs and had domiciled in the lungs, with downstream effects on the heart. He fit the stereotypical case of cor-pulmonale-right sided heart failure from severe lung disease. The facts of the case suddenly began to make a lot of sense to me. I thus had a better understanding of why the patient had progressed so rapidly with his disease course with a fatal outcome.
I realized later that all my prior apprehension about performing the autopsy had been replaced by an interesting curiosity to find out more about his disease. My initial trepidation about performing that autopsy was quickly replaced by a determination to answer the “why” question. I became more involved and present with the procedure that by the time we left the autopsy suite, I thought I had learned something new that day.
That experience of being able to solve a clinical puzzle from autopsy findings made a huge impact on me. Therefore, the role of pathology and laboratory medicine in the advancement of medicine and patient care can never be overstated.
-Evi Abada, MD, MS is a Resident Physician in anatomic and clinical pathology at the Wayne State University School of Medicine/Detroit Medical Center in Michigan. She earned her Masters of Science in International Health Policy and Management from Brandeis University in Massachusetts, and is a global health advocate. Dr. Abada has been appointed to serve on the ASCP’s Resident’s Council and was named one of ASCP’S 40 under Forty honorees for the year 2020. You can follow her on twitter @EviAbadaMD.
The SARS-CoV-2 virus continues to cause increased infections and deaths around the world with considerable impact on clinical and laboratory medicine communities. Meanwhile, medical students and the medical community are also undertaking the yearly tribulation of residency interview season. Following the May announcement by the Coalition for Physician Accountability’s Work Group on Medical Students,1 the 2020 interview season will be entirely conducted utilizing virtual interviews. In pointed response to this change in format, residency programs rapidly scrambled to bolster websites, increase their social media presence, add virtual tours and prepare for the virtual interview format prior to the start of interview season. Now, at the midpoint of interview season, it is evident that some burdens of traditional on-site interviews are indeed being alleviated. Whether or not online resident socials and virtual tours can sufficiently substitute for all aspects of on-site visits and if the promise of increased spread of geographic and cultural diversity can be realized remains to be accurately assessed. The survival of the virtual format may even depend on this assessment.
The average cost of traditional on-site pathology interviews has continued to increase for medical students from a per person average of $3400 in 2015 to $4000 in 2020.2 Much of this expense comes from travel/transportation while some pathology programs provided accommodations. Additionally, interview season required about 20 total days away from medical school. To cover these expenses, about half (49%) of medical students borrow money for interviews . Not surprisingly, the majority of them agree that travel (79%) and lodging (65%) are overly burdensome components of interview season.2 Beyond accounting, the salient impact of these time and financial investments is that they were influencing the majority (58%) of interview decisions.
While the rising time and financial burdens of traditional on-site residency interviews were well-known and there was and continues to be a myriad of ideas3 on how to best address these concerns and the match overall, a small burgeoning literature on virtual resident interviews was available prior to the pandemic that showed promise for addressing these concerns.4,5 That is, in the 2020 – 2021 residency interview season, medical students are estimated to spend about 3.5 hours on an average virtual interview day instead of the 8 hour day of a traditional interview and through the elimination of travel time they may spend 7 less days on the interview trail. Thus, the cost of interviewing is also projected to be skeletonized to that of necessary professional clothing and computer hardware. Additional promising data from this small body of research suggests that 85% of virtual interviewees were satisfied with their understanding of the program and their ability to present themselves to residency programs.6 Furthermore, the fact that the residency program’s rank list showed no significant impact based on whether candidates interviewed virtually or in-person suggests that residency programs may feel capable of fairly assessing candidates.7
Beyond time and financial savings for pathology residency applicants and the assessment of candidates by residency programs and vice versa, the measurability of additional outcomes may be critical to the continuation of virtual resident interviews. In particular, there is great interest in online social events and interview day resident panels as a sufficient substitute for the naturally evolving casual conversations that occur during the dinners, lunches and tours available with on-site visits. Also, whether or not these socials combined with interviews with a small subset of faculty can accurately portray a pathology residency program’s culture. In prior surveys that compared in-person, virtual or a combined approach to interviews, candidates always favored in-person assessment when given the choice. The present circumstance will perhaps be the best attempt at an unbiased assessment of the perception of culture through virtual interviews. Last but not least, given the turbulent nature of race relations and culture in the United States over the last year combined with the ability of applicants to virtually interview without travel or financial restrictions, it will be absolutely critical to understand if virtual interviews portend to increase the spread of geographic and cultural diversity among applicants to pathology residency programs. That is, if current trends in resident recruitment can be altered from the current rate of 40 – 60% intraregional resident matriculation or whether the needs of financial and family assistance and/or intraregional familiarity are insurmountable.8 For if the potential for greater diversity is attainable in a significant manner that can be perpetuated into the future, it will be hard to argue for a return to the traditional format. That said, there will likely be bias in the data as an increasing number of pathology residency programs have heard the call to arms and are marching towards diversity, inclusion and equity through greater promotion, recruitment and retention efforts.9
In a tumultuous year that has included race relations reminiscent of the Civil Rights Era combined with a total number of worldwide pandemic deaths similar to the 1957 or 1968 influenza pandemics, medicine continues its steady progression toward improved healthcare and education for all. Following the May 2020 recommendations to implement virtual residency interviews, pathology residency programs moved expeditiously to bolster their websites, increase their social media presence, add virtual tours and prepare for the virtual interview format. Amid this tumult, the virtual interview format has already served to lessen the burdens of time and cost while also serving the practical needs of interview assessments for both medical students and residency programs. Yet, only time and methodical assessment will tell if the virtual interview format eliminates the impact of these burdens on residency decisions, allows both parties to adequately assess cultural fit and if the format and its advantages are here to stay. Regardless, it is imperative that the emphasis on diversity, inclusion and equity remains irrespective of future format.
References
The Coalition for Physician Accountability’s Work Group on Medical Students in the Class of 2021 Moving Across Institutions for Post Graduate Training Final Report and Recommendations for Medical Education Institutions of LCME-Accredited, U.S. Osteopathic, and Non-U.S. Medical School Applicants.
Pourmand, A., Lee, H., Fair, M., Maloney, K. & Caggiula, A. Feasibility and usability of tele-interview for medical residency interview. Western Journal of Emergency Medicine 19, 80–86 (2018).
Hammoud, M. M., Andrews, J. & Skochelak, S. E. Improving the Residency Application and Selection Process: An Optional Early Result Acceptance Program. JAMA – Journal of the American Medical Association 323, 503–504 (2020).
Chandler, N. M., Litz, C. N., Chang, H. L. & Danielson, P. D. Efficacy of Videoconference Interviews in the Pediatric Surgery Match. J. Surg. Educ. 76, 420–426 (2019).
Vining, C. C. et al. Virtual Surgical Fellowship Recruitment During COVID-19 and Its Implications for Resident/Fellow Recruitment in the Future. Ann. Surg. Oncol. 1 (2020). doi:10.1245/s10434-020-08623-2
Healy, W. L. & Bedair, H. Videoconference Interviews for an Adult Reconstruction Fellowship: Lessons Learned. Journal of Bone and Joint Surgery – American Volume 99, E114 (2017).
Vadi, M. G. et al. Comparison of web-based and face-to-face interviews for application to an anesthesiology training program: a pilot study. Int. J. Med. Educ. 7, 102–108 (2016).
Shappell, C. N., Farnan, J. M., McConville, J. F. & Martin, S. K. Geographic Trends for United States Allopathic Seniors Participating in the Residency Match: a Descriptive Analysis. J. Gen. Intern. Med. 34, 179–181 (2019).
Ware, A. D. et al. The “Race” Toward Diversity, Inclusion, and Equity in Pathology: The Johns Hopkins Experience. Acad. Pathol. 6, (2019).
-Josh Klonoski, MD, PhD, is a chief resident at the University of Utah, Salt Lake City, Utah, with a focus in neuroinfectious disease and global health. He has completed the first year of a neuropathology fellowship (out of sequence) and is in his final year of an anatomical and clinical pathology residency. Dr. Klonoski will return to the second neuropathology fellowship year in 2021 – 2022 and apply for a mentored clinical scientist research career development award (K08). The focus of his laboratory research is influenza and active projects include flu pneumonia, super-infections, encephalitis and oncolytic virotherapy.
Last time we talked a bit about what exactly pathology and laboratory medicine training looks like—a much-needed peek behind the curtain, if you ask me. This time, I’d like to discuss something that’s been challenging me way before I started working as a resident: something that’s made both clinical medicine and academic collaboration difficult (to say the least). I talk a lot about how medicine works best when we all come together and contribute our expertise from disciplines across the board for the sake of improving patient care; I even talk about how, too often, people don’t hear the messages coming through from their colleagues and that negatively impacts our field. We’re all guilty of it, some more than others. And in the last few weeks, I’ve been nearly deaf to my colleagues.
No literally, this article is about my Meniere’s disease and how those of us with invisible illness(es) can really impact the collaborative spirit of medical science. Trigger warning: frustration, anxiety, and pathology (mine, specifically). So here’s a reminder that everyone you meet and work alongside has issues they’re dealing or struggling with. And, to those outside of medicine, remember doctors are people too. Dr. Jena Martin, a dermatopathologist I admire and follow on social media recently promoted the topic #DoctorsArePatientsToo—she writes, shares, and promotes fantastic topics on social media and I suggest you check her content out at @jenamartinmd.
Image 1. By poet and patient Ronny Allen who was diagnosed with neuroendocrine cancer in July of 2010. He is passionate about education and awareness and advocates for patients everywhere. (Source: ronnyallan.com)
So, I’ve got a couple things to explain here. Come with me on a journey through the inner ear and out into the reaches of global pandemics!
An MD with MD
So what is Meniere’s disease (MD) and why does it deserve its own article? Well, first of all, it doesn’t deserve anything, it’s a garbage disorder with dumb symptoms and I’m only using it/myself as an example to highlight the struggle for other people working in medicine who also deal with invisible problems. But, since you asked…
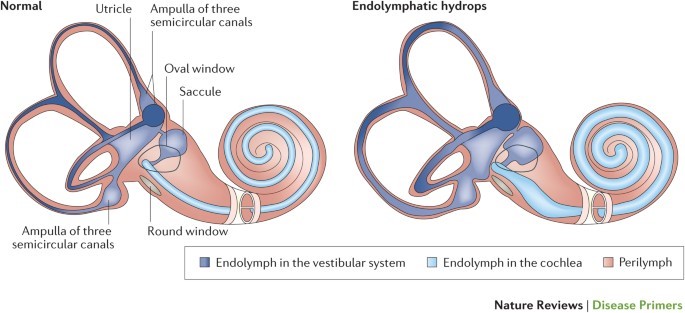
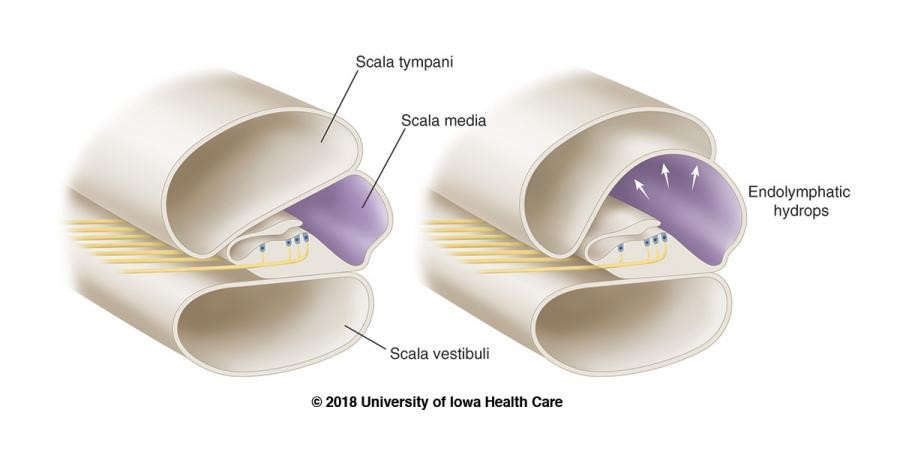
Without belaboring any detail, Meniere’s is a somewhat understood inner ear disorder in which the potassium-rich, sound-signal inducing endolymphatic fluid builds up in the inner ear. This causes the organ of Corti (Image 2b) to swell up, along with the rest of the cochlea, the vestibule, and the fluid containing parts of the inner ear (Image 2a). The super pressurized vestibular-cochlear balloon is in a confined bone-space so what happens often is that small fistulas form, mixing the endo and perilymphatic fluids causing all sorts of problems not limited to but including: aural fullness, deafness, ridiculous multi-tonal/pulsatile tinnitus, distorted hearing, frequency loss, imbalance, severe peripheral/rotational vertigo lasting for hours, occasional tachycardia, anxiety, and more! There is no cure; therapy is symptom-dependent and purely aimed at management and mitigation of fulminant hearing loss. Did I mention that most times it’s one-sided, but mine is bilateral? Fun. I’ll be donating my ears to science and/or the garbage in some odd decades from now…
Image 2a and 2b. Dr. Strange-sound, or how I learned to stop worrying and love endolymph. Inner workings of the inner ear, note the marked swelling of fluid (hydrops) associated with the disease state compared to normal. (Sources: Nature and University of Iowa)
Let me put it this way: Over time, I will lose a stark majority of my natural hearing. I will continue to get occasional vertigo attacks, and related symptoms, until the disease (eventually…hopefully?) “burns out” and has no more capacity to damage any more already-damaged inner ear hair cells. I’ll probably get hearing aids or cochlear implants. (Whatever, I’ve always wanted to be half bionic…) I’ll probably keep a cane with me most times. (Dr. House anyone? Right? Or I could keep a sword in there? Legal issues?) But it’s not the long term that bothers me most. It’s the flare-ups. This disorder has periods of acute attacks, periods of mixed-symptom flare ups, and periods of remission. I can handle the remissions, hah. I can even handle the acute episodes—I’ve had great ENT colleagues and discovered great medication plans to manage the attacks. So that leaves the flares, which inspire writings like this one obviously. I wear my hearing aid, but it’s minimally helpful, and I practice patience until my hearing returns. But that isn’t as easy as it sounds, especially when you’re a working resident MD!
The Sound of Silence
Technically, I haven’t experienced true silence since 2015. And, during flare-ups, most of my hearing is almost entirely replaced by the only sound my ears can accommodate and attenuate for which is their own local vasculature. If Edgar Allan Poe could see me know…telltale ears, anyone? Let me paint a picture of what I’m hearing right now on a pretty rough flare-up day, at work, at my desk in the resident room. I’m drinking tea, I can’t hear myself swallowing. I monitor my heartrate on my watch and it’s about 80-90 beats per minute, in my ear, all day, whoosh-whoosh. The pot of tea the robots have been making, whistles non-stop, 24/7. The mosquitoes I’ve been writing about for years now on Lablogatory never leave my cochlea. Cool, on top of that I’ve got hearing loss and aural fullness that makes me not able to hear low-volume pages/phone rings (vibrate mode FTW) and most talking—especially behind masks. (I’ll get to that in a second.) If I turn my hearing aid too high, there’s a squeaky feedback explosion, not to mention the occasional adjustments for quiet and/or super loud talkers, yikes. Just overall, me no hear too good right now. Basically if hearing was reading text, the printer is out of toner and the paper is provolone cheese. And the cheese is on fire. Opa!
Image 3. My flaming-cheese metaphor captured, a xerox copy being made by a waiter in Chicago’s Greektown, the original home of the flaming-cheese saganaki dish. (Source: Chicago Sun Times)
Global Pandemic? Sounds Pretty Bad…
The SARS-CoV-2 pandemic has created very interesting situations at work and elsewhere, all over the globe. People are utilizing the most effective measure against spreading the illness: which up to this point is the non-pharmaceutical intervention of social distancing. Nearly every single person working today, in any field, doing anything, reading this blog, has been a part of email chains, and conference calls, and …. sigh… Zoom meetings. I know, I know… there are pro’s and con’s to this but consider these three points for those with #InvisibleIlnesses:
Working from home has liberated chronically ill folks
Okay so I have an intermittent vestibular/cochlear disorder. What about folks with chronic pain, or Lyme-sequelae, or brain fog, or any other host of hurdles before they can jump onto a video call. Pretend you’re sick at home with a (non-COVID) viral bug. Can you imagine how nice it would feel to have hot tea, your medications, your cat, your spouse, and (most importantly) your couch/bed/TV nearby? Within reason, you’ve just given people who would have needed to excuse themselves a way to participate productively!
2. Imagine having a supplementary PowerPoint or chat box during conversations IRL
One of my specific challenges is missing a word here, a sentence there, and not being able to catch up in a conversation. Usually it ends up with my smile-nodding through to the next topic or checkpoint, but in the current age of virtual meetings I can un-obtrusively ask “What was that last bit?” without seeming like I tuned out. And bonus: I can often get a text translation of a point or two I might have missed from keen colleagues aware of my AV troubles. I can only imagine what it would be like for those more permanently affected.
3. The explosion of inclusivity and accessibility is remarkable
Speaking of which, the number of videos and presentations I am now seeing with closed captions/text supplements are astounding. Usually when I’m in the midst of a flare, I have to check out from audiovisual stimuli and stick to reading only. This can be quite the challenge for work sometimes. But with chat boxes and videos with captions, I feel like I can catch right up. Now, I said I’m only like this part of the time, so for those that have felt excluded and marginalized I’m happy to say that inclusivity and accessibility are growing. I’ve often thought that the importance of a news story or press conference could be measured by the presence or absence of a sign-language interpreter. Over 100 press conferences by Governor Andrew Cuomo in NY, or in Chicago, Illinois with Governor JB Pritzker and Mayor Lori Lightfoot, and each of them was accompanied by ASL accessibility. Fantastic! Just look at one of many efforts online, like @ProjectHearing on Instagram, which promotes the advancement of these topics every day!
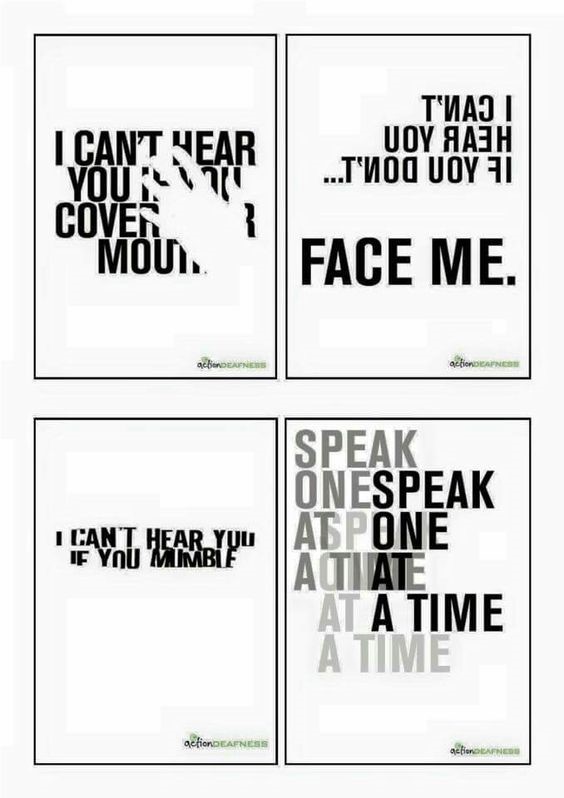
Image 4. “Deaf people problems” is a meme collection I’ve seen over and over on social media. Awareness, check. Clear message, check. Super creative visual way to demonstrate a better version of my flaming cheese analogy, super check.Image 5. Don’t tell anyone, but sometimes, I’m literally stuck on mute. Video conferences might become the new normal. If they are, remember to speak clearly and keep things out of the way between your talking and your microphone. Consider captions for videos and make nice PowerPoints. Please, haha.
Your Lips Move but I Can’t Hear What You’re Saying
So why, when the world is literally aflame with a viral pandemic, am I drudging you through my rant against my inner ears? And why did I just commit the travesty of endorsing zoom calls and captions (I know some people hate captions—you came for a movie not a book, I get it).
Well this whole topic illustrates to big things about working both as a physician and as a person with an invisible condition. First, like most things in medicine, to achieve success you have to adapt, improvise, and overcome. Solving patients’ problems and advocating for their best outcomes takes a little finessing of the system sometimes, you’ve got to do the same thing for yourself. Second, since doctors are patients too, its okay to ask for help. I matched with some of the best residents I’ve ever met, and they’ve offered whatever they can in helping manage my flares during work. This includes anything from extra emails/group text notification chains, forwarding pages, translating video call jumbled audio, etc. They are the best!
Meniere’s has been a challenge to me for a couple of years now, and it’s something I deal with. We’ll call it a character-building attribute. But I genuinely did worry about how this was going to affect me during residency—I had my fair share of hard days in med school basic sciences, and plenty of attending wave-offs when I simply couldn’t hear on 4:00am rounds (yeah I’m looking at you OBGYN and Surgery…). But, it’s been good.
Image 6. Not a new Arnold Schwarzenegger movie, these are real and they’re incredible. Clear window facemasks are such a relief for me. I give ones to all the folks I work with and my life just got easier.
And more than just good, another resident actually has a more permanent hearing impairment and two desks over from mine are the embedded hearing interpreters provided by the department. They’ve been so friendly and provided my with so many resources that I couldn’t be more appreciative. Not only do I have another resident to confide hearing-related rants to, but I also have a department that cares enough to create a supportive and accessible environment. One of the best things they’ve provided: MASKS WITH WINDOWS! Because since this damn pandemic started, I can’t read lips anymore! I didn’t realize how much I depended on visual lip-reads to confirm my hearing that it’s been a learning curve to say the least. Imagine being mostly deaf, your hearing aid not helping much, and looking into a multi-headed scope while your attending lectures on what you’re looking at. An otherwise impossible situation, but my friends and colleagues find ways to make it work because when one person excels, we all do. I’ve been able to continue working, learning, and collaborating thanks to considerations for invisible illnesses like mine.
Consider your colleagues, what can you do to make sure they feel that their needs are met since #DoctorsArePatientsToo? See you next time!
-Constantine E. Kanakis MD, MSc, MLS (ASCP)CM is a new first year resident physician in the Pathology and Laboratory Medicine Department at Loyola University Medical Center in Chicago with interests in hematopathology, transfusion medicine, bioethics, public health, and graphic medicine. His posts focus on the broader issues important to the practice of clinical laboratory medicine and their applications to global/public health, outreach/education, and advancing medical science. He is actively involved in public health and education, advocating for visibility and advancement of pathology and lab medicine. Watch his TEDx talk entitled “Unrecognizable Medicine” and follow him on Twitter @CEKanakisMD.
Thanks for reading my piece last month on liquid biopsies. And, as a side note, there is a growing number of awesome quality content and posts from pandemic response, to inclusion, alongside COVID and case-studies so subscribe, share, and add this page to your bookmarks—STAT! Lablogatory has been a fantastic platform to share and learn so much in this past year, I could barely keep up!
Or super-STAT if you’re one of those people…but hey, that language belongs to all of us! Lab professionals, nurses, scientists, and doctors alike. And this month, I just want to take a quick moment to celebrate a milestone.
I’m officially a resident physician/trainee, medical post-graduate! (There was confetti falling just now on my end, not sure about yours, but work with me here.) It’s just one of those life-goals that feels great when you get there. But there’s a lot more to it than it seems…if I told you being a pathology resident means sacrificing early adulthood, amassing soul-crushing debt, and explaining to your peers and colleagues what it is exactly you do and why you also bear the moniker of “physician,” you’d delete that bookmarked webpage faster than I can make you scroll through this thing.
(oh good you’re still here!)
All that said, I’ve got to say: it’s worth every single bit of it. Times a million. But I really did mention some red flags that, were we discussing any other work environment, would make you definitely think twice before committing 5-10 years of your life. Furthermore, as a PGY-1 in pathology, I could stand next to any other patient-facing PGY-1 colleague (read: intern) and they wouldn’t have the faintest about what I actually do. Listen, the “lifestyle” specialty, generally 9-5er, no weekend, no 24hr call isn’t something I’m shy to celebrate, but it’s not the whole story. I’ve matched and started learning and working at a great institution with great faculty, mentors, and other residents/fellows. Bottom line: I’m more than a little happy about where I’m at professionally.
Image 1. Most path & lab med residents get cubicle-style desks to spend time reading, prepping, writing, learning, and previewing cases between responsibilities in sign-outs, tumor boards, or OR/gross room work. Most of my non-path friends don’t like this. It makes me very happy.
So, to my non-pathology friends, what is it exactly that I do during my residency training while you might be busy rounding, managing glucose levels, triaging cases, putting orders in—you know, regular intern stuff *shudders* … (pathology trainees don’t have an intern year, we jump right into the specialty and go for 3-4 straight on through). Like most of you I have a transition period where I get acclimated to the workload and patterns of my specific residency, but sans-anno-interna, I’ve got lots of work ahead to climb the steep learning curve that med school pathology merely skims.
What does non-patient-facing mean, exactly?
Well, without an intern year you jump right into what most path residents go into which is a 4-year combined anatomic and clinical pathology (AP/CP) track. You immediately begin training in all the fields in pathology. They include surgical pathology (of various sub specialties like head-and-neck, gynecologic, gastrointestinal, thoracic, neuro, etc.—think surgery, then add pathology), autopsy training, dermatopathology, cytopathology, hematopathology, transfusion medicine, clinical chemistry, microbiology, hemostasis and coagulopathy, pediatric pathology, forensic pathology, molecular, training as a laboratory director, and much, much more. Each of these services has a workload which is usually comprised of cases from biopsies and grossed specimens for histologic analysis (anatomic pathology) or the ongoing maintenance and advancement of clinical diagnostic testing through laboratory methods and management of staff/resources (clinical pathology).
Image 2. August 2019 Issue of The Pathologist magazine. Pathologists, medical students, microscopes, you get it…Specifically, that’s one of my mentors (and now faculty) Dr. Kamran Mirza and (then) medical student Austin McHenry discussing the critical role pathology plays in every circle of medical care.
When I say “non-patient facing” this means that the majority of that work is not done in 1-on-1 settings with patients in a clinic or hospital floor. It is done ancillary to their clinical experience whereby pathology attendings manage the simultaneous training of residents and processing of case sign outs for rapid and accurate diagnostic output for our patient-facing colleagues. For example, while a patient, their family, and doctor are discussing and managing symptoms related to a possible cancer diagnosis. The pathologists are examining microscopic behavior of the cancer-in-question’s cells and adding immunohistochemical testing and molecular analyses to identify, stage, and prognosticate that cancer. Returning information about what it is and what can be done back to the patient-facing clinician, who can then best-translate a tailored approach for their patient. Old-timey medical texts would often refer to the pathologist as the “doctors’ doctor,” and I’m not here to hate on that haha. My clinical friends and readers might feel forlorn now at the prospect of 4 years of medical school training to “just look into a microscope all day?” Well, for some folks in path it means a lot more than that, every slide is a patient. So we care just as much as if they were right opposite our desk. But that’s not all we do…
(More on that in a minute.)
So What Do You Do?
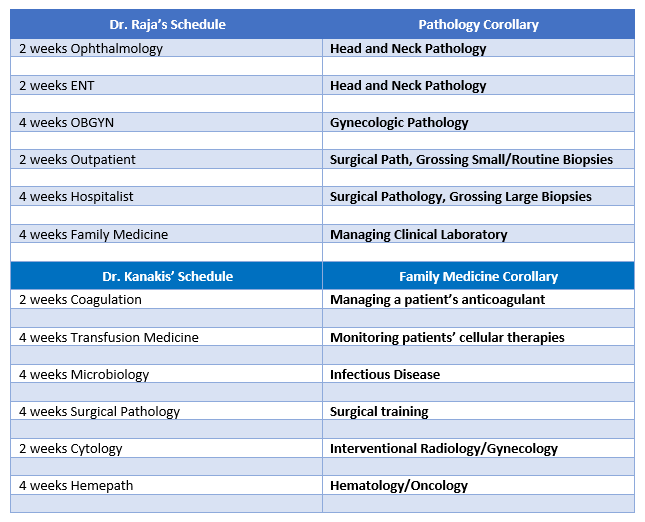
Okay, there are lot of words in path that might act as a barrier to understanding the common ground between me and …let’s say a colleague and friend in Family Medicine. So for the purposes of transparency here’s my friend from medical school Dr. Danash Raja and how a small part of his schedule and my schedule aren’t so different…
Image 3. Dr. Raja is from Alaska, and now works as a resident physician in Family Medicine in Eu Claire, Wisconsin! Alaska! Look at this graduation photo!
On both sides of this table are clinicians managing their patients and ensuring the best possible outcomes. Both sides are deeply vested in intensive hours of training, procedural experience, evidence-based best-practices from the literature, and ongoing continuing education.
Image 4. They see me grossin’, they hatin’…A lot of surgical pathology and microscopy in general revolves around understanding the gross layout of a specimen and its orientation before it becomes a thin microscope slide. As a junior pathology resident, we spend a lot of time up near the OR. Critical skill for a crucial foundation of knowledge.
Literally the biggest differences:
In pathology, I get my own desk space and I need it! I’ve got to start amassing a physical and digital library to supplement the next 4-6 years of subspecialty training for the eventual day when a colleague will see me in an elevator and expect concise, thorough, and actionable material to inform their clinical management from the pathologic diagnosis.
Pathology residents and clinical residents both take “call” except Dr. Raja has to pull grueling 24-hour+ shifts and stay in the hospital for the duration, and I answered a page about a transfusion reaction from a grocery store once.
When a patient thinks about the person who helped them find out what kind of cancer they had and what treatment to begin, they’ll probably think of Dr. Raja or someone patient-facing in Heme/Onc—but I’m working on this, every day!
Bottom line: I’m as important as he is, and he is as important as I am. Our work is what really matters, and what really connects us as clinical colleagues. It’s all about patients, remember? But I’m more than happy to be the pathologist to his patient-facing, diabetes-managing, vaccine-giving, life-improving super hero doctor!
You Never See Patients?
We’re back on this. Remember how I said looking into microscopes isn’t all we do? Okay, well it’s not. And if you’re lucky enough to have matched to as awesome of a place as I did, then you know what I’m talking about. If you’ve read some of my pieces, you know full well my passion in pathology lies in Hematopathology and Transfusion Medicine. I like to sit right on the fence between AP and CP, and mostly look at the green grass on the CP yard. This month, I’ve been on service for Transfusion Medicine and let me tell you about the few weeks…
Image 5. Dr. Kimberly Sanford, ASCP leadership and Director of Transfusion Medicine at VCU was highlighted in The Pathologist magazine for her work outside the laboratory seeing patients every day, and encouraging residents to do the same and do what pathologists do best: enrich and improve the channels of communication so patients better understand their conditions and the medical process.
I, a pathologist trainee, resident physician, under the supervision of two attending physician pathologists have been seeing and following up on patients nearly every day. Gasp! No, I’m not part of some backwards resident exchange program (because OMG how dangerous haha), no I’m not lost, no I’m not being overly gunnery, that’s it, that’s the Tweet. Seriously, it’s just part of the service. Larger academic hospitals with robust clinical blood bank services often have apheresis clinics and I find myself working exactly there. Blood bank/Transfusion Medicine is one of those subspecialties where patient contact is part of the routine. At some institutions, I’ve been a part of some pathology-led teams that procure the bone marrow aspirates from their patients in Hemepath service, or conducted their own fine needle aspirations for cytology service, or dermpath services that operate in clinics alongside their dermatology colleagues—I’ve even been working on frozen sections and surgical path grossing when called into an operating room to discuss methods and approach for biopsy! There were patients at every turn, all with pathologists on the front line! Dr. Syed T. Hoda (@01sth02 on Twitter) from NYU Langone often says, “Person FIRST, doctor SECOND, specialist THIRD.” And trust him, he’s a bone and soft tissue pathologist that left the lab and went to the floors to help clinical staff when overwhelmed during the peak of the COVID crisis in NYC. So for my dual-interests, I would say I’d expect to see quite a bit of patients in my future practice.
Image 6. My awesome co-residents! (Left-to-right): me, Dr. Elnaz Panah, Dr. Aayushma Regmi, and Dr. Sandra Haddad—you’re going to hear more about them, don’t worry.
So, would I pick pathology again? Uh, yeah! Without a single hesitation. Every day at work I am reminded that I am at the right place, with the right co-residents, the right faculty and mentorship, and the right environment to train and hone my future skills for a career that lines up exactly with what I want to do.If you’re interested about the intersections between clinical medicine and pathology, and want to learn more about “patient-facing pathology” keep an eye out during the 2020 ASCP Annual Meeting for a talk by yours truly as part of a panel discussion on communicating directly with patients! Register now! Free for members.
See you next time!
BONUS: did you notice that I referenced The Pathologist magazine a bit in this post, well it’s because they named me to their Pathology Power List for 2020! An exclusive, international list of 80 professionals in the field of pathology and laboratory medicine who contribute and advance the profession every day! I was highlighted for my active social media work and my response to the early COVID pandemic in Manhattan, NY.
-Constantine E. Kanakis MD, MSc, MLS (ASCP)CM is a new first year resident physician in the Pathology and Laboratory Medicine Department at Loyola University Medical Center in Chicago with interests in hematopathology, transfusion medicine, bioethics, public health, and graphic medicine. His posts focus on the broader issues important to the practice of clinical laboratory medicine and their applications to global/public health, outreach/education, and advancing medical science. He is actively involved in public health and education, advocating for visibility and advancement of pathology and lab medicine. Watch his TEDx talk entitled “Unrecognizable Medicine” and follow him on Twitter @CEKanakisMD.
After a lot
of positive responses and sharing on social media, my article last month got lots of people talking about
annual meetings and how great they are for networking, learning, and advancing
our profession. Not too long after the ASCP Annual Meeting in Phoenix, I was
back in my Manhattan apartment working on my speech and graphics for a real
life TEDx session hosted at my medical school.
Let’s pause
here: if you either haven’t heard of the TED/TEDx brand or if you binge watch
their 18 minute videos and want more links to watch now, now, now!
TED
is a non-profit organization whose mission is to share “ideas worth spreading.”
They’re about 35 years old and based in NYC stateside, and Vancouver in Canada.
Basically, over the last few decades they hold conferences at those flagship
sites called “TED talks” where selected speakers present on a myriad of topics.
TEDx conferences are officially licensed but off-site events which operate
under TED protocol and guidelines. There have even been spin-off conferences
like TED MED, which focus solely on healthcare.
Image 1. What’s a TEDx talk? Basically, an off-site, officially sanctioned, “idea sharing” conference.
Some of the students at AUC School of Medicine, organized such a conference with official TED licensing and recruited me to join their list of speakers to deliver talks on their chosen theme: resilience. Officially called TEDxAUCMed, this conference included community members, students, artists, activists, and more discussing the human capacity for resilience in ways not commonly discussed. “Weathering the Storm” was the official event title, as the school located in the island nation of St. Maarten displays daily resilience especially since being hit by Hurricane Irma in 2016. Among their list of incredible speakers, I was humbled to be included! I titled my talk “Unrecognizable Medicine” and wanted to deliver a talk to students, clinicians, and those of us in medicine witnessing first-hand a tidal wave of new technologies and paradigms that redefine the way we discuss health. Oh, and since I’m a huge fan of #GraphicMedicine more and more each day, I hit that hashtag hard and decided to illustrate my whole talk!
Image 2. Title Card from my TEDx talk.
So what did I talk about, exactly…and what’s the big deal? I’m not going to re-hash my presentation for you in text—that’d be boring, and I’m obviously going to put a link at the bottom for you to watch it yourself. I got you, lab fam! But essentially, what I set up was a three-tiered template to assess and navigate that tidal wave of tech. Tools, skills, and strengths—three things inherent to the practice of medicine in any specialty.
Image 3. Red back-ligting. So intense. Thanks for coming to my TEDx Talk, literally!
There are
untapped topics in medicine which are looming over the horizon. As medicine
continues to evolve and change, the problems we face and the needs we must meet
will become moving targets. New specialties will emerge, and new technologies
will replace centuries old tools we cling to today. A shift in thinking is both
proactive and healthy in a profession that mandates our commitment to
preserving health and quality of life. I have spent years battling stereotypes
in medicine and hope to challenge the fabric that places individuals in
professional or academic boxes. Fresh first-years at some schools are already using
point-of-care ultrasounds (POCUSes) instead of stethoscopes—which
student sounds like they have better info on morning rounds, a student who
maybe kinda-sorta heard some non-descript murmur, or a mini-pocket
echocardiogram with an ejection fraction of 45%? Stereotypes have too long
shaped the way students choose specialties, equating some areas to colloquial
high school cliques! No offense to orthopedics or dermatology. Troponins used to be something you
could hang your white coat on, but not anymore. What do you do with a new 5th
generation Trop of 39 with a delta of 18? ACS or acute MI? Cancer therapy is
exploding with personalized treatments being added every day! Any student right
now would impress their heme/onc attending on rounds if they suggested PDL-1
and other immunotherapy testing for patients with newly diagnosed lung cancers.
*Deep breath*
Ok. My point
is, tomorrow’s medicine is going to have a lot of different therapies, tools,
and even vocabulary that schools may never catch up with. How do you prepare
for this explosion of knowledge? You look to yourself to take an inventory of
your strengths and use those to guide your clinical sails. Addressing
stereotypes head-on, learning on the spot, dealing with complex identities in
your patients, and always practicing with compassion will lend itself to
staying ahead and staying fulfilled.
Image 4. If you’re drawing cartoons of pathologists for an educational series, you probably make them look like you. Or in this case me, I guess. Keep an eye out for my #PathDoodles on social media!
Pretty heavy stuff right? But there’s something else that caught my attention in reflection on the TEDx talk… I’ve searched the TED library of videos, and while there are plenty of doctors, scientists, and pioneers in research discussing medical ideas, I haven’t seen any medical laboratory scientists. If you find any, please correct me. But, as I understand it, it’s just me. And that’s something special.
Image 5. My wife and I check-in for rehearsal at the TEDxAUCMed conference in sunny St. Maarten.
There’s a
culture shift in our profession, and a lot of us are talking about it.
Pathology and laboratory medicine are stepping out from behind the healthcare
curtain and asserting itself as a champion for patients, truth, and the
importance of data-driven medicine. Not only do I talk to groups of folks every
time I get a stage, but I use social media to reach clinicians and patients!
Yes, I’m one of few medical students-turned-residency applicants who didn’t
change their name to hide their online presence for the winter. But instead of
a secret twitter hibernation, I’ve used social media as a tool to network,
engage, and connect.
One of my
favorite new projects is something I call #PathDoodles where I break down the
aspects of pathology and some specialty topics for those outside of medicine
(and sometimes just outside our profession). I’ve already covered things like
“what is pathology?” and the importance of autopsies, the role of medical
laboratory scientists, and I continue to add more regularly!
Image 6. One of a growing list of #PathDoodles.
There’s a
culture shift in our profession, and a lot of us are talking about it.
Pathology and laboratory medicine are stepping out from behind the healthcare
curtain and asserting itself as a champion for patients, truth, and the
importance of data-driven medicine. Not only do I talk to groups of folks every
time I get a stage, but I use social media to reach clinicians and patients!
Yes, I’m one of few medical students-turned-residency applicants who didn’t
change their name to hide their online presence for the winter. But instead of
a secret twitter hibernation, I’ve used social media as a tool to network,
engage, and connect.
One of my
favorite new projects is something I call #PathDoodles where I break down the
aspects of pathology and some specialty topics for those outside of medicine
(and sometimes just outside our profession). I’ve already covered things like
“what is pathology?” and the importance of autopsies, the role of medical
laboratory scientists, and I continue to add more regularly!
Follow me on Twitter (@CEKanakisMD) and check out my TEDx talk:
My talk begins at 5:00:00. Enjoy!
–Constantine E. Kanakis MD, MSc, MLS (ASCP)CM completed his BS at Loyola University Chicago and his MS at Rush University. He writes about experiences through medical school through the lens of a medical lab scientist with interests in hematopathology, molecular, bioethics, transfusion medicine, and graphic medicine. He is currently a 2020 AP/CP Residency Applicant and actively involved in public health and education, advocating for visibility and advancement of pathology and lab medicine. Follow him on Twitter @CEKanakisMD
The patient is a 43 year old woman who experienced chest congestion and presented to her local physicians office. A chest X-ray was ordered and demonstrated a lung abnormality. A follow-up CT scan confirmed a 1.9 cm smoothly marginated nodule in the upper lobe with no adenopathy and a normal liver and adrenal glands. The nodule was mildly hypermetabolic on PET scan. A bronchoscopy was performed, which was non-diagnostic. Two subsequent CT scans demonstrated no change in the size of the nodule. Overall, the patient feels well and denies cough, hemoptysis, dyspnea on exertion, and weight loss. Due to the suspicion of cancer, the patient has decided to undergo a lung lobectomy.
Diagnosis
Received in the Surgical Pathology lab for
intraoperative consultation is a 30.0 x 7.2 x 2.2 cm lung lobectomy specimen. There
is an attached 6.2 cm staple line, which is removed and the subjacent resection
margin is inked blue. The entire pleural surface is inked black. The specimen
is sectioned revealing a 2.1 x 1.7 x 1.0 cm white-tan, firm, round nodule that
is 0.5 cm from the blue inked resection margin and 0.2 cm from the black inked
pleural surface. The remainder of the specimen is composed of red-tan, spongy,
grossly unremarkable lung parenchyma without nodules or other lesions.
Photographs of the specimen are taken (Figure 1). A representative section of
the nodule is submitted for frozen section and read out as “diagnosis
deferred”. Representative sections of the specimen are submitted as follows:
A1FS: Frozen
section remnant
A2-A7:
Nodule, entirely submitted
A8-A10: Grossly unremarkable lung parenchyma
Immunohistochemical stains show the epithelial cells in the lesion to be positive for CK7, TTF-1, and surfactant proteins A and B which supports these cells to be type 2 pneumocytes (all controls are appropriate). Based on the immunohistochemical stains and routine H&E slides, the case was signed out as a sclerosing pneumocytoma
Image 1. Gross presentation of the well-defined, round sclerosing pneumocytoma.
Discussion
Sclerosing pneumocytoma (SP) is a rare, benign pulmonary
tumor that was first described in 1956 as a vascular tumor, but has since been
found to be of primitive respiratory epithelium origin. In the past, SP has
also been referred to as sclerosing hemangioma, pneumocytoma, and papillary
pneumocytoma, but the 2015 World Health Organization classification of lung
tumors states that the agreed upon term for this tumor should be a sclerosing
pneumocytoma. SP is commonly seen in middle aged adults, with a female to male
ratio of 5:1. There is no racial bias. Patients are usually asymptomatic, with
the tumor incidentally found on screening chest radiographs. If the patient was
to present with any symptoms, they would usually include a cough, hemoptysis
and chest pain. Radiographically, SP appears as a solitary, well-defined,
homogenous nodule along the periphery of the lung.
Grossly, most SPs appear as a solitary, firm, well-circumscribed,
yellow-tan mass generally arising along the periphery of the lung. The majority
of these tumors appear within the lung parenchyma, but there have been cases
reported of endobronchial and pleural based SP tumors. Multifocal unilateral
tumors and bilateral tumors are uncommon.
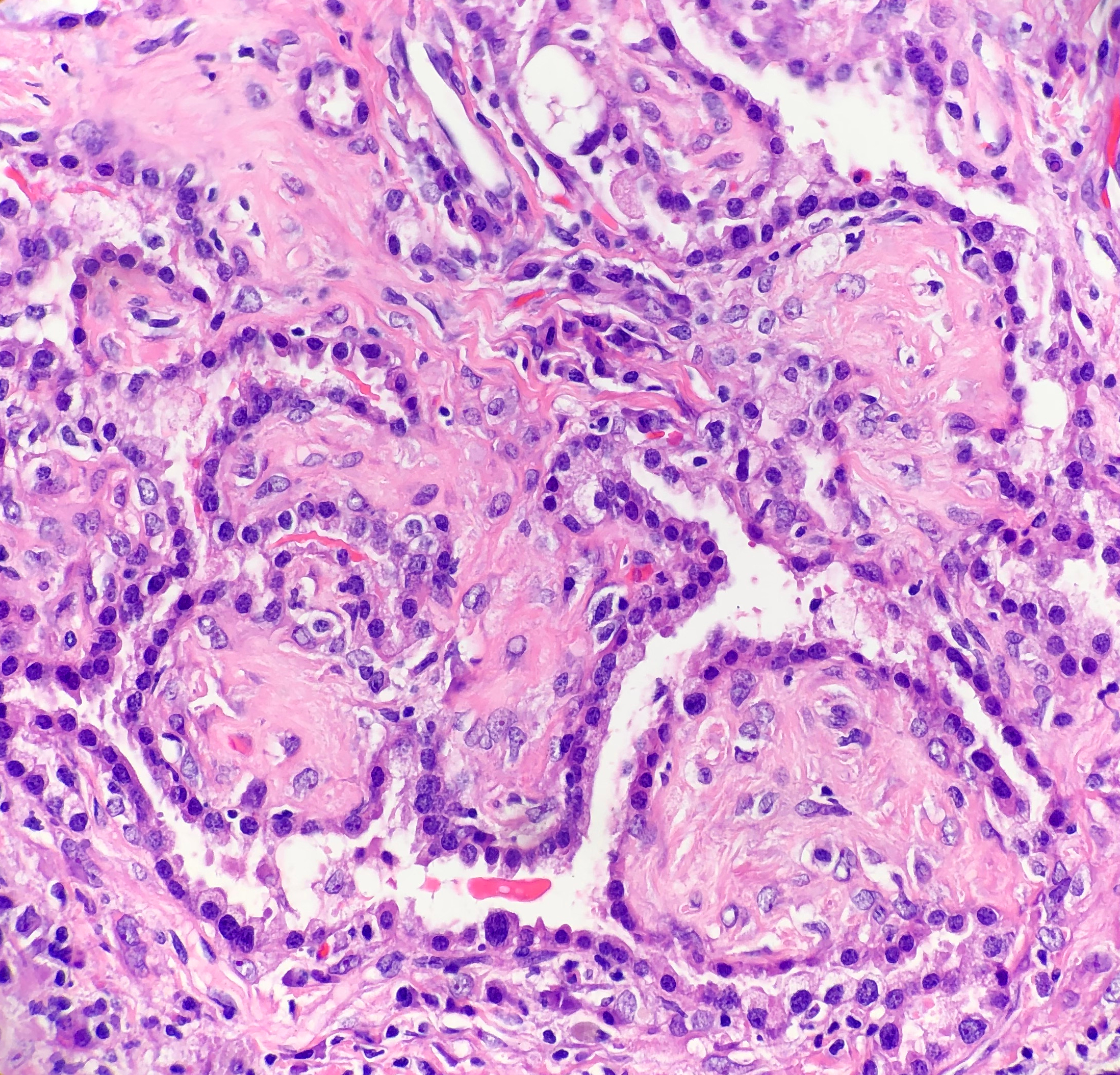
Histologically, SP consists of two epithelial cell types:
surface cells and round cells. Surface cells are cuboidal, resembling type II
pneumocytes, with finely stippled nuclear chromatin, indistinct nuclei,
occasional nuclear grooves, and inclusions. The stromal round cells will have bland
oval nuclei with coarse chromatin and eosinophilic cytoplasm (Figure 2). Both
the surface cells and round cells will have a low mitotic rate, but can have
moderate to marked nuclear atypia. Ciliated bronchial epithelium is often
identified in the tumor. There are four architectural patterns identified
within SP: papillary, sclerotic, solid and hemorrhagic, with over 90% of SPs
displaying three of the patterns, and all of the tumors containing at least two
of the patterns.
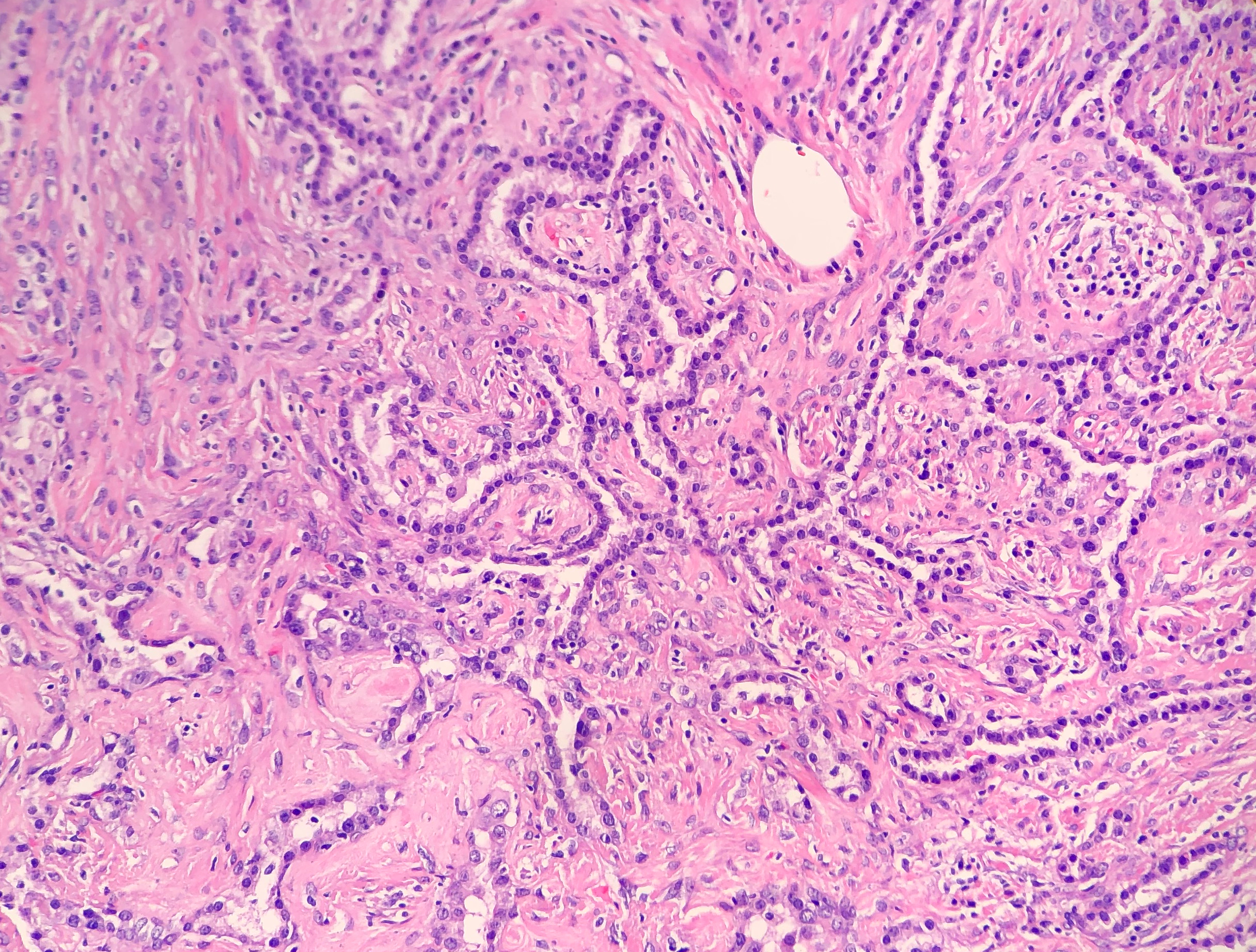
Papillary pattern: Complex papillae composed of surface cells covering a stroma of round cells
Sclerotic pattern: Papillae containing hyalinized collagen, either in solid areas or along the periphery of hemorrhagic areas (Figure 3)
Solid pattern: Sheets of round cells bordered by surface cells
Hemorrhagic pattern: Large blood filled spaces
Image 2. Photomicrograph demonstrating the cuboidal surface cells and round stromal cells.Image 3. Photomicrograph of the papillary and sclerotic architectural patterns.
Immunohistochemical stains can be helpful in the diagnosis of
SP, with both the surface cells and round cells exhibiting expression of
thyroid transcription factor 1 (TTF-1) and epithelial membrane antigen (EMA). It
should be noted that TTF-1 is also used for the diagnosis of pulmonary
adenocarcinoma, increasing the risk of misdiagnosing SP. The surface cells will
also express both pancytokeratin (AE1/AE3) and Napsin A, with the round cells
being negative for AE1/AE3, but having a variable expression of cytokeratin 7
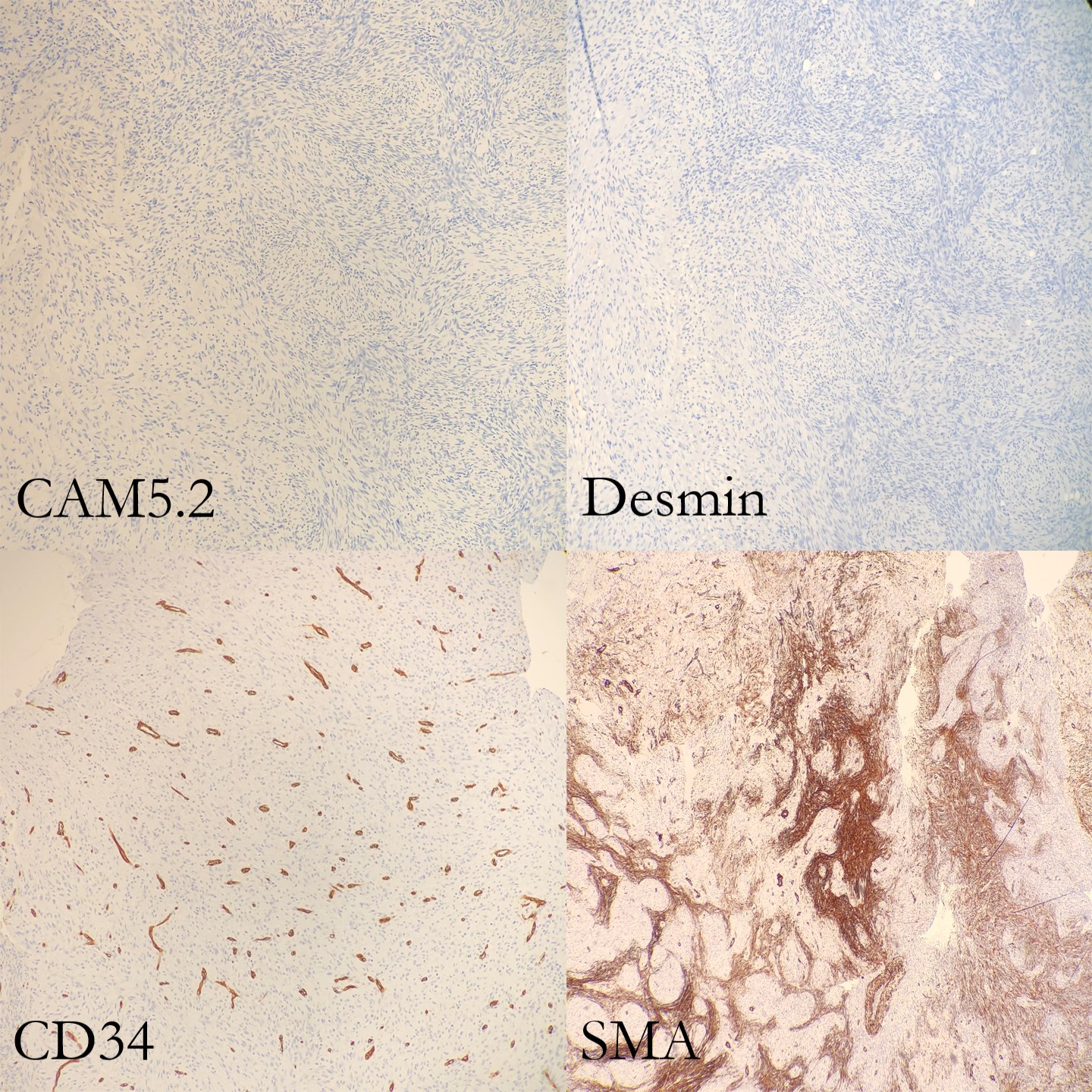
and the low molecular weight cytokeratin (CAM 5.2). Molecular pathology has demonstrated
a frequent loss of heterozygosity at 5q, 10q and 9p, and an allelic loses at
p16 in the surface and rounds cells. Although the immunohistochemical stains
and molecular pathology results can be very helpful, diagnosis of a SP is still
largely based on routine H&E slides showing the two epithelial cell types
and four architectural patterns.
Electron microscopy will show abundant lamellar bodies
similar to those in type II pneumocytes in the surface cells. Round cells will
lack the lamellar bodies and instead will contain variably-sized electron-dense
bodies that have been thought to represent the different stages of lamellar
body maturation.
The differential diagnosis for SP includes a variety of
benign and malignant neoplasms, which can be difficult to distinguish on
cytology, small biopsies and intraoperative consultations. The cytologic
features include moderate to high cellularity with a bloody background and
foamy macrophages, occasional nuclear pleomorphism in the round cells, absent
mitotic figures, and occasional necrosis with cholesterol clefts and
calcifications. In the case of small biopsies, making a diagnosis of SP can be
difficult if the papillary pattern is highly prevalent without one of the other
three patterns present. With intraoperative consultations, the frozen section
artifact can make it difficult to appreciate the two epithelial cell types or
the four architectural patterns. The gross examination, as well as the
radiographic findings of a well-circumscribed tumor can help point the
Pathologist to favoring a benign neoplasm over a malignant one. The benign
neoplasms that should be considered in the differential diagnosis include:
Clear cell tumor, which will have clear cells
with scant stroma, thin-walled vessels and a strong expression of HMB-45
Pulmonary hamartoma, which will have a
combination of cartilage, myxoid stroma, adipose tissue and trapped respiratory
epithelium
Hemangiomas, which are rare in the lung, and will
lack epithelial cells and contain either a cavernous or capillary morphology
The malignant neoplasms that should be considered in the
differential diagnosis include:
Bronchioalveolar carcinoma, which can have a
papillary pattern, but will not contain the two epithelial cell types and
combination of the four architectural patterns
Metastatic papillary thyroid carcinoma, which is
distinguished from SP by the presence of the characteristic Orphan Annie nuclei
Metastatic renal cell carcinoma, which will
contain nuclear atypia and striking vascularity
Carcinoid, which will contain organoid and
ribbon-like growth patterns
Currently, with the benign nature of SP, surgical excision is
the preferred treatment choice to cure the patient. There have been cases
reported of lymph node metastasis and recurrence, but neither of these appear
to effect the prognosis. This just helps to highlight the need for a
multidisciplinary approach to this benign tumor.
References
Hisson E, Rao R. Pneumocytoma (sclerosing
hemangioma), a Potential Pitfall. Diagn
Cytopathol. 2017;45(8):744-749
Keylock JB, Galvin JR, Franks TJ. Sclerosing
Hemangioma of the Lung. Arch Pathol Lab
Med. 2009;133(5):820-825.
Travis WD, Brambilla E, Nicholson AG, et al. The
2015 World Health Organization Classification of Lung Tumors: Impact of
Genetic, Clinical and Radiologic Advances Since the 2004 Classification. J Thorac Oncol. 2015;10(9):1243-1260.
-Cory Nash is a board certified Pathologists’ Assistant,
specializing in surgical and gross pathology. He currently works as a
Pathologists’ Assistant at the University of Chicago Medical Center. His
job involves the macroscopic examination, dissection and tissue
submission of surgical specimens, ranging from biopsies to multi-organ
resections. Cory has a special interest in head and neck pathology, as
well as bone and soft tissue pathology. Cory can be followed on twitter
at @iplaywithorgans.
The patient is a 3
year old male with no significant past medical history who presented to the ED
with left lower extremity pain for 24 hours after falling while playing with
family members. The patient’s mother was present at bedside providing the history,
but was not present at the time of the fall. It is unclear how the patient
injured his ankle, but family members noticed the child grabbing his ankle and
suspected that he may have twisted it. After the fall, the patient was unable/unwilling
to ambulate on the ankle. There is no history of fractures or cancer.
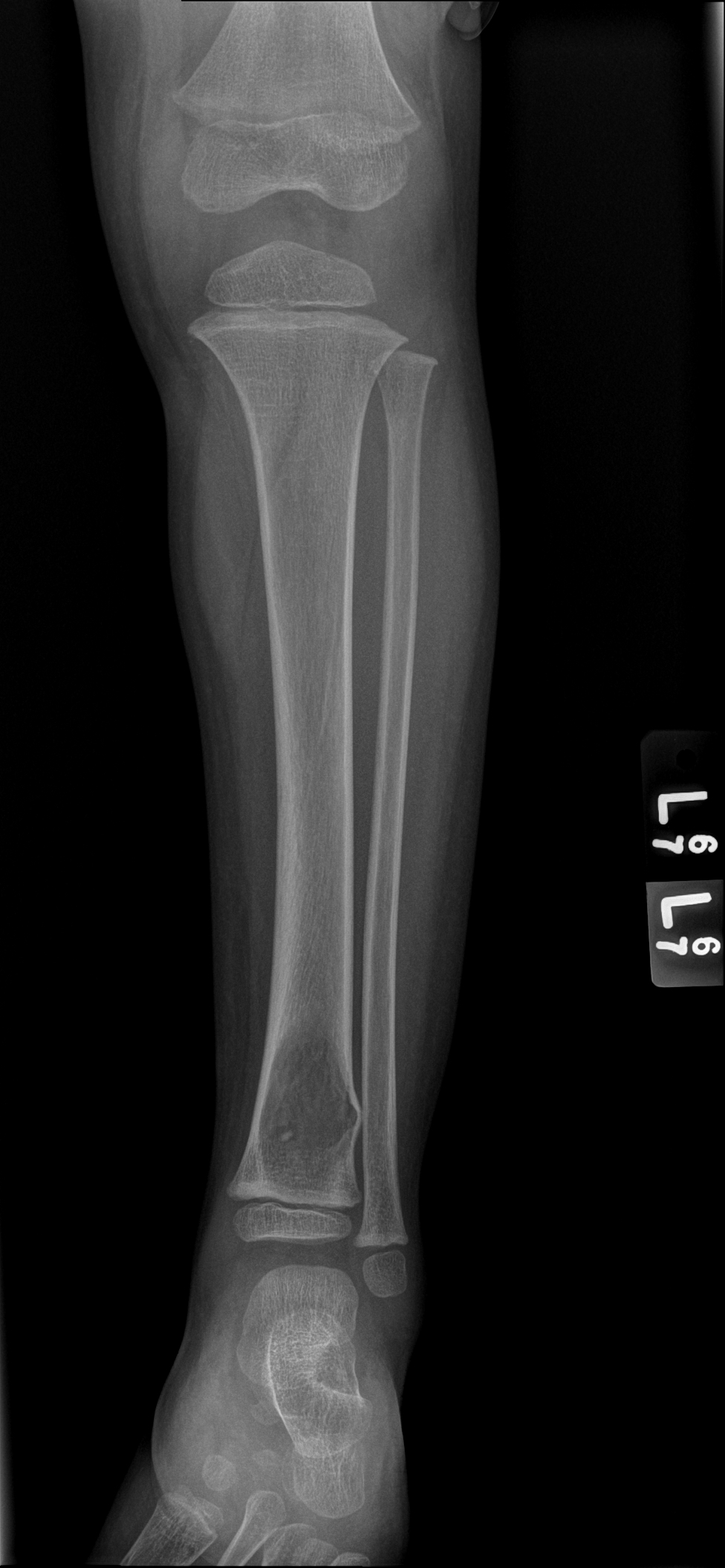
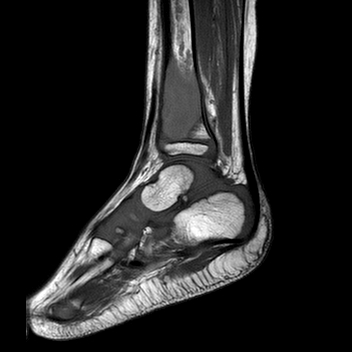
An x-ray and
subsequent MRI were ordered of the ankle which demonstrated an expansile lytic
lesion involving the metaphysis of the distal tibia measuring approximately 3.4
x 2.2 cm (Figure 1 and 2). The margins of this lesion are indistinct, and there
is cortical irregularity at the anterior and lateral aspect of the distal metaphysis
of the tibia, likely representing a pathologic fracture. The differential
diagnosis includes infection, aneurysmal bone cyst, nonossifying fibroma,
osteoblastoma and histiocytosis.
The patient and family
then followed up with Orthopedics, who proceeded to perform a biopsy of the
lytic lesion in order to determine the nature of the lesion. The results are
below.
Figure 1. Xray of the distal tibia demonstrating the lesion.
Figure 2. MRI demonstrating the lytic lesion involving the metaphysis of the distal tibia.
Diagnosis
Received fresh for intraoperative consultation is a 1.1 x 0.6
x 0.5 cm aggregate of white-tan soft tissue fragments. Half of the tissue
fragments are frozen and read out as “spindle cell proliferation.
Consideration of low-grade vasoformative lesion. Defer to permanent,” with
3 pathologists consulting on the diagnosis. The remainder of the tissue not
submitted for frozen section, as well as the entirety of a second container from
the same lesion, is submitted for routine processing.
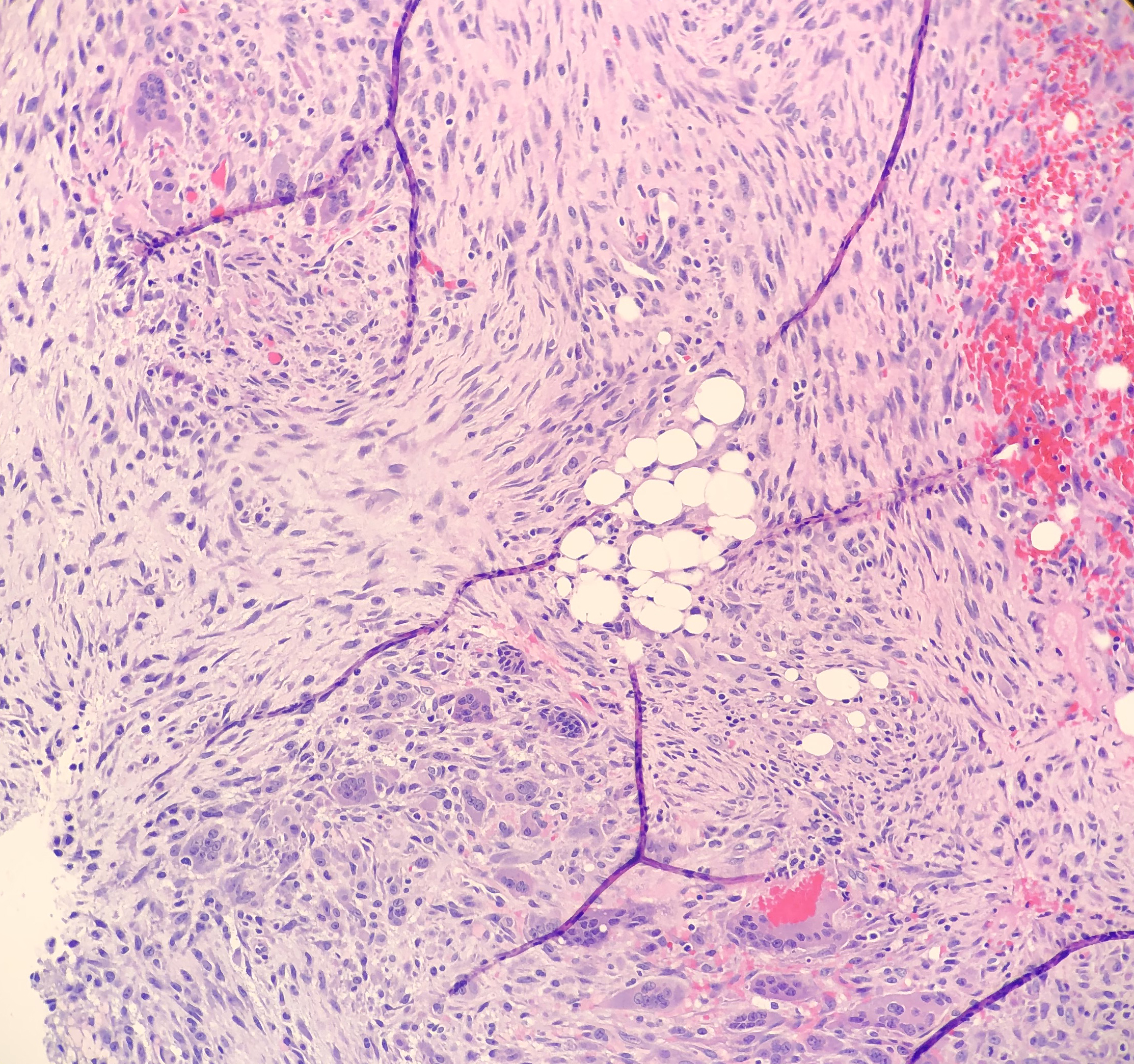
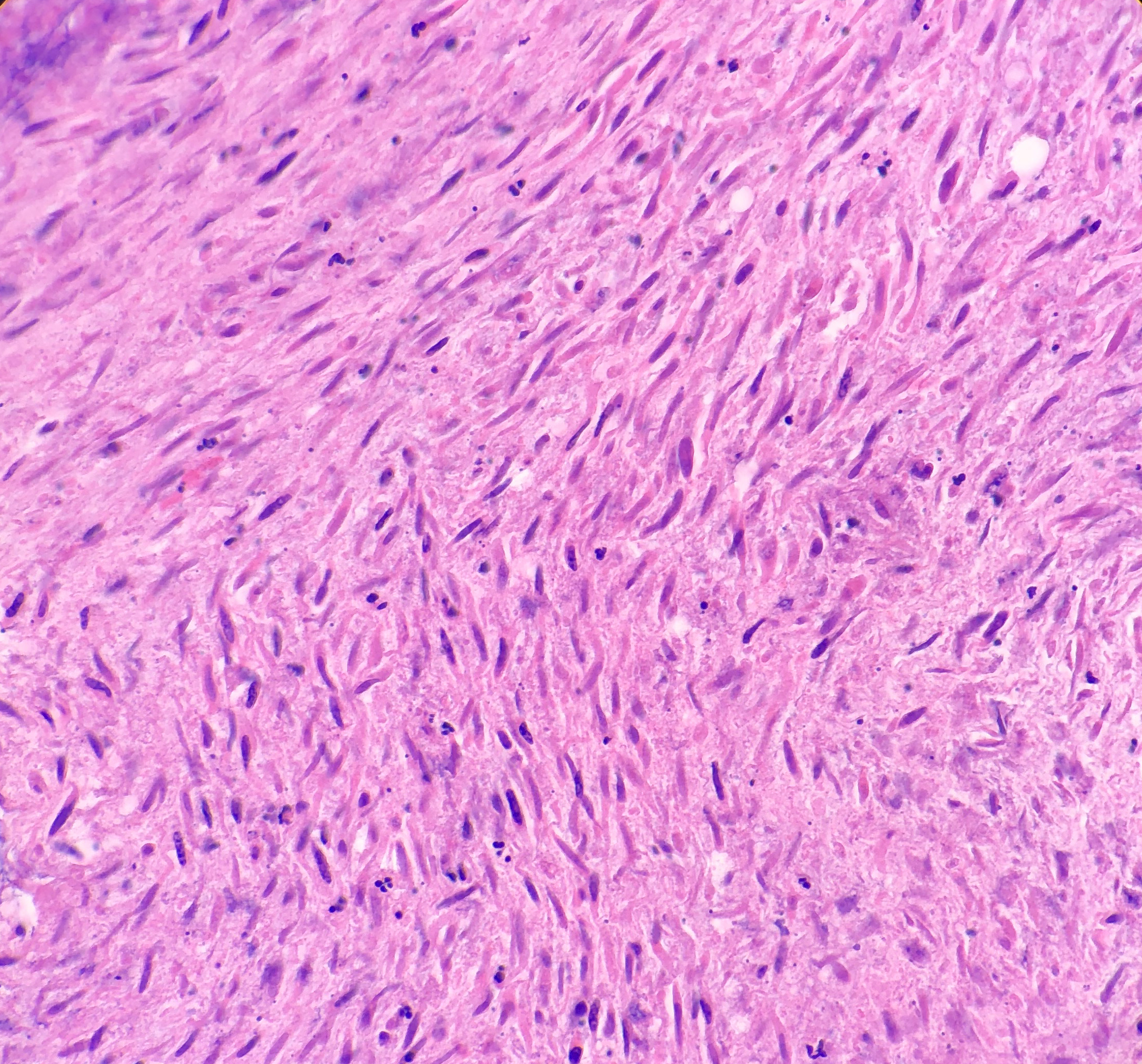
On microscopy, the biopsies demonstrate a moderately cellular
proliferation of fasciculated spindle cells in a collagenous to myxoid stroma.
Nuclei are predominantly oval with variably fine to granular chromatin. Many
cells have moderate amounts of tapering eosinophilic cytoplasm, resembling
strap cells. Inflammatory cells and osteoclast-like giant cells are admixed
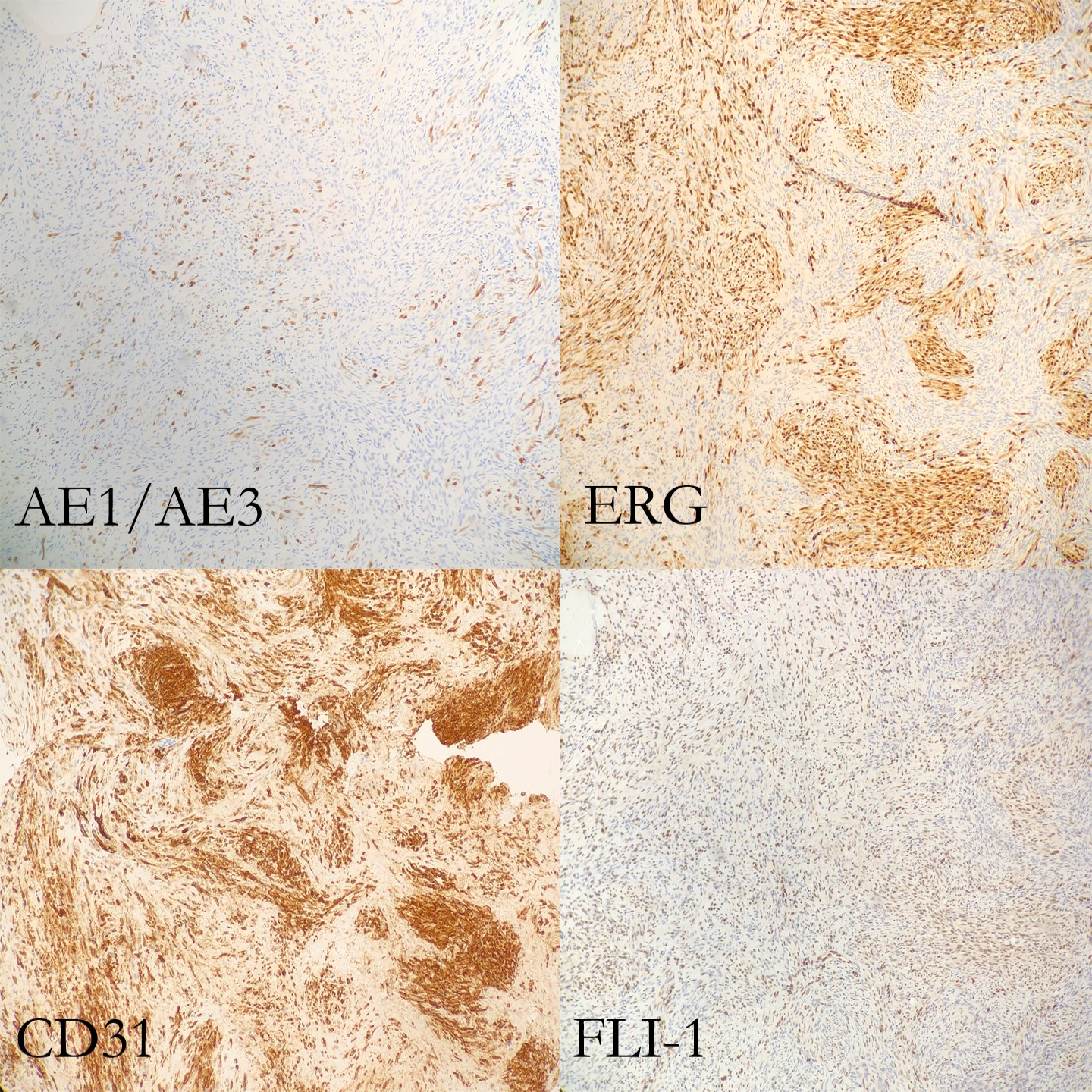
(Figure 3 and 4). Immunohistochemical stains demonstrate lesional spindle cells
to be positive for CD31, ERG, and FLI1. AE1/AE3 and CAM5.2 highlight rare
lesional spindle cells. SMA stains some stellate spindle cells, favored to
represent associated myofibroblasts. Desmin, MDM2, CDK4, ALK, and S100 are negative
in plump lesional cells (Figure 5 and 6). Overall, the features are consistent
with pseudomyogenic hemangioendothelioma, a rare vascular tumor. Although more
commonly present in soft tissue, primary bone cases have been reported. These
neoplasms have some risk for local recurrence, but only rarely distant
metastasis. A portion of tissue was sent to the University of Nebraska Medical
Center to evaluate for a characteristic gene rearrangement (SERPINE1-FOSB) that
is present in at least a subset of pseudomyogenic hemangioendotheliomas. This
was negative.
The lesion was then curettaged by the
surgical team.The patient and his family had two follow up office visits with
the Orthopedics department. The first one, a week after surgery, was
unremarkable. The second visit, two weeks after surgery, was notable for the
patient developing a cutaneous rash on both arms and chest. Due to literature
citing that these tumors generally arise in the soft tissue, the clinician
suggested that the patient and family follow up with pediatric dermatology to
ensure that this new rash is not related to the pseudomyogenic hemangioendothelioma.
Unfortunately due to insurance, the patient and family had to see a dermatologist
at a different institution, and no further visits have taken place.
Figure 3. Photomicrograph of the strap-like cells with tapering eosinophilic cytoplasm , and osteoclast-like giant cells. Figure 4. Higher power photomicrograph demonstrating the appearance of the strap-like cells with tapering eosinophilic cytoplasmFigure 4.
Discussion
Pseudomyogenic hemangioendothelioma (PHE) is a rare vascular
tumor that most commonly arises in the skin and soft tissues of the
extremities. It is usually multifocal, appearing in multiple tissue planes,
such as the mucosa, dermis, subcutis and skeletal muscle, in a variety of
different anatomic sites. Although even less common, PHE can also involve bone
(such as this case). PHE has a male predilection, typically appearing in the second
to fourth decades of life. Of the most common symptoms that the patient
presents with, pain appears to top the list, although it should be stated that
only about half of the patients experience pain.
Grossly, skin and soft tissue PHE tumors appear firm,
ill-defined and gray-white. When they involve bone, they appear as multiple
discrete, pink-tan to dark brown hemorrhagic tumors with surrounding sclerosis,
ranging from 0.1 to 6.5 cm in greatest dimension.
Histologically, PHE demonstrates plump spindle and rhabdomyoblast-like
cells with densely eosinophilic cytoplasm that grows in sheets and fascicles. The
cells can be mistaken as rhabdomyoblasts because of the eosinophilic cytoplasm
that pushes the nucleus to the periphery of the cell. Immunohistochemical studies are very helpful in order to
determine a diagnosis of PHE. AE1/AE3, ERG, FLI-1 and CD31 are positive,
whereas CD34, desmin and S100 are negative. Karyotyping has revealed a
fusion of genes SERPINE1-FOSB that
corresponds to the recurrent translocation t(7;19)(q22;q13). In this case, the SERPINE1-FOSBgene rearrangement was negative, but could possibly be due to
a variant fusion gene.
Making a histologic
diagnosis can be difficult for a Pathologist, due to the wide variety of
differential diagnoses that will need to be excluded first.
The differential diagnosis
for a cutaneous tumor includes:
Cellular benign
fibrous histiocytoma (lacks rhabdomyoblast-like cells and neutrophilic
infiltrates, contains mitotic figures, and is negative for cytokeratin and CD31)
Spindle cell
squamous cell carcinoma (usually in sun-damaged skin, with nuclear atypia and
negative endothelial markers)
Epithelioid
sarcoma (negative INI1, positive EMA and CD34, and a nodular architecture with
central necrosis and more nuclear atypia)
The differential diagnosis
for soft tissue tumors include:
Epithelioid
sarcoma (see above)
Epithelioid
hemangioendothelioma (usually intracytoplasmic vacuoles, positive CD34 and
CAMTA1, and a t(1;3)(p36.3;q25) translocation resulting in WWTR1-CAMTA1 gene fusion)
Epithelioid
angiosarcoma (vasoformative architecture with sheet-like pattern, nuclear
atypia, high nuclear grade, frequent mitosis and irregular vascular channels)
The differential diagnosis for bone tumors
includes:
Giant cell tumor
(lacks rhabdomyoblast-like cells and fascicles of spindle cells)
Osteoblastoma
(lacks rhabdomyoblast-like cells and fascicles of spindle cells)
In a study by Inyang et al,
when PHE involved bone, imaging would demonstrate multiple to innumerable
discontinuous tumors throughout the affected bone, involving the cortex and/or
medullary cavity of the epiphysis, metaphysis, or diaphysis. On x-ray and
computed tomography, the lesions appeared as well circumscribed, lobulated and
lytic, with a sclerotic rim on some of the lesions. On magnetic resonance
imaging, T1-weighted images would appear dark, and T2-weighted images would
appear hyperintense.
PHE has a tendency to recur
locally, but rarely develops distant metastases. Since PHE presents as a
multifocal disease and can be easily confused for a distant metastasis, care
needs to be taken to ensure that a diagnosis of PHE is not overlooked.
Surgical ablation and
excision is the standard treatment for a patient with PHE, with a few cases
noted of patients being treated with radiotherapy and/or adjuvant chemotherapy,
in addition to surgery. Everolimus and sirolimus have recently been found to be
effective in cases of patient with PHE that had metastatic and relapsing multifocal
PHE.
Figure 5. Immunohistochemical stains (part 1 of 2)Figure 6. Immunohistochemical stains (part 2 of 2)
References
Hornick JL, Fletcher CDM. “Pseudomyogenic Hemangioendothelioma:
A Distinctive, Often Multicentric Tumor With Indolent Behavior.” Am J Surg
Pathol. 2011; 35: 190201.
Inyang A, et al. “Primary Pseudomyogenic Hemangioendothelioma
of Bone.” Am J Surg Pathol. 2016; 40: 587598.
Pradhan D. “Pseudomyogenic hemangioendothelioma
of skin, bone and soft tissue; a clinicopathological, immunohistochemical, and
fluorescence in situ hybridization study.” Hum Pathol. 2018; 71: 126134.
Sugita S, Hirano H, Kikuchi N, et al. Diagnostic utility of
FOSB immunohistochemistry in pseudomyogenic hemangioendothelioma and its histological
mimics. Diagn Pathol. 2016;11(1):75. Published 2016 Aug 11.
doi:10.1186/s13000-016-0530-2
-Cory Nash is a board certified Pathologists’ Assistant,
specializing in surgical and gross pathology. He currently works as a
Pathologists’ Assistant at the University of Chicago Medical Center. His
job involves the macroscopic examination, dissection and tissue
submission of surgical specimens, ranging from biopsies to multi-organ
resections. Cory has a special interest in head and neck pathology, as
well as bone and soft tissue pathology. Cory can be followed on twitter
at @iplaywithorgans.
The patient is a 2.5 year old male who is being evaluated for
a liver transplant versus biliary diversion surgery. The patient was born at 2
kilograms and went home with mom one week after birth. The patient was
readmitted back to the hospital for evaluation of jaundice and since then the patient
has been intermittently hospitalized for episodes of worsening jaundice,
acholic stools, scleral icterus, and pruritus. At 5 months of age, the patient
was diagnosed with progressive familial intrahepatic cholestasis, type 2, and
was placed on the liver transplant list. As a result of the liver failure, the
patient has developed coagulopathy, hypocalcemia resulting in seizures, and
pruritus. The family history is significant for no known congenital
liver diseases.
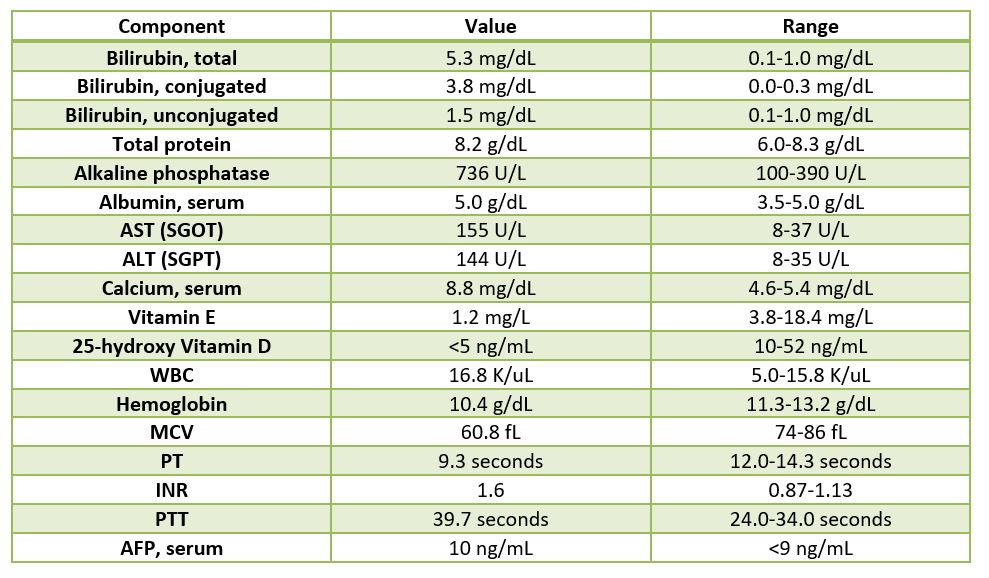
Table 1. Pertinent lab findings.
The father was worked up for living donation and was found
to be a suitable donor, and is donating the left lateral segment of his liver.
Diagnosis
Received
in the Surgical Pathology laboratory is a 700 gm, 23.5 x 14.5 x 3.5 cm
explanted liver with an attached 4.5 x 1.2 x 0.4 cm gallbladder. The liver
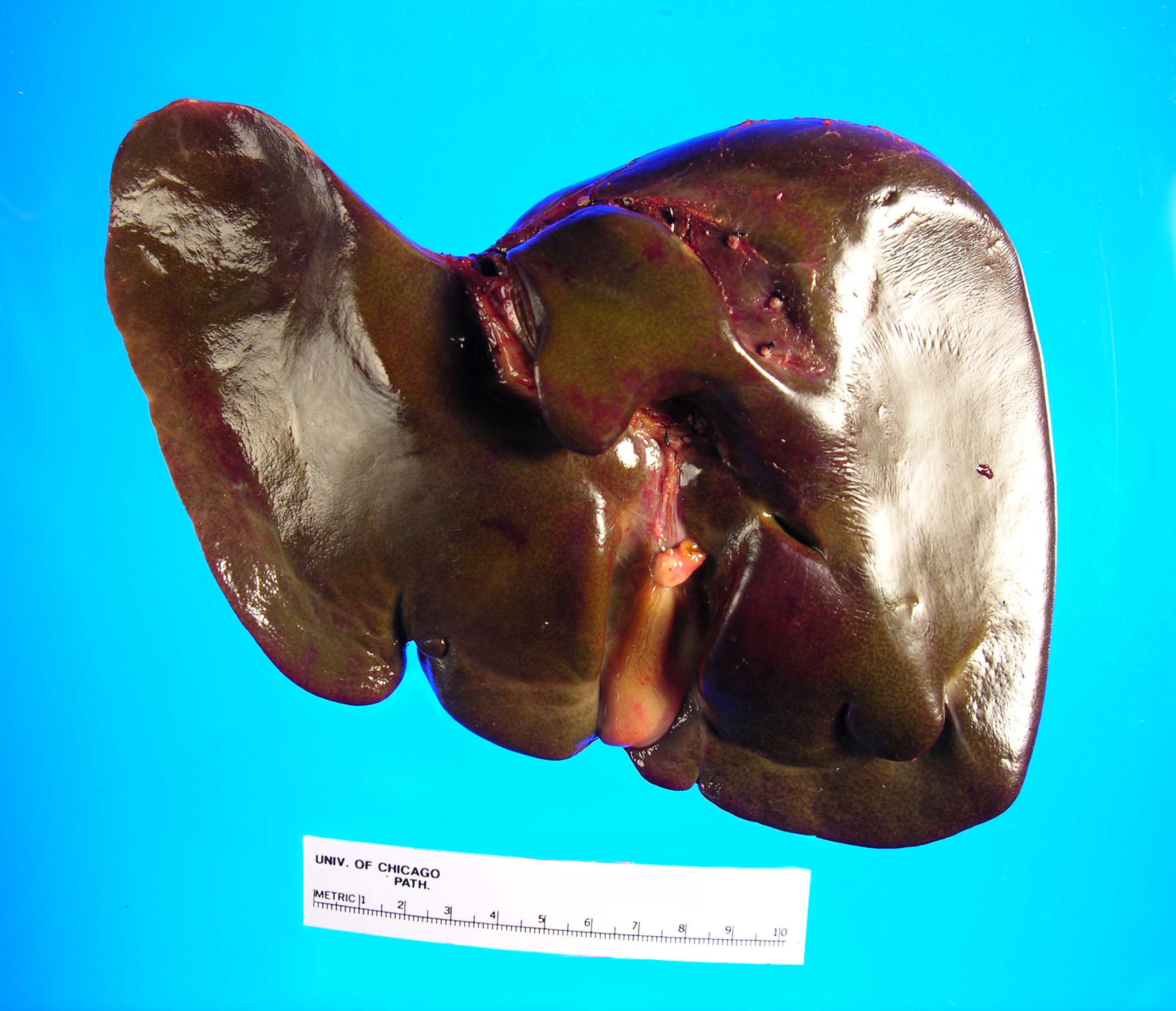
specimen has a smooth, green-red liver capsule without any grossly identifiable
nodules or lesions (Image 1). The gallbladder has a yellow-pink external
surface and is opened to reveal a 1.5 x 0.7 x 0.4 cm dark brown stone with a
small amount of brown-yellow bile fluid. The liver is sectioned to reveal a
smooth green-red cut surface (Image 2). No lesions are identified and minimal
hilar structures are included with the specimen. Portions of the specimen have
been taken for electron microscopy and frozen for future diagnostic purposes.
Submitted sections include:
Cassette
1 and 2: Hilar structures
Cassettes
3-15: Representative sections of
liver parenchyma
Cassette
16: representative section of
gallbladder
Image 1. Posterior aspect of green-tinged liverImage 2. Cut section of liver
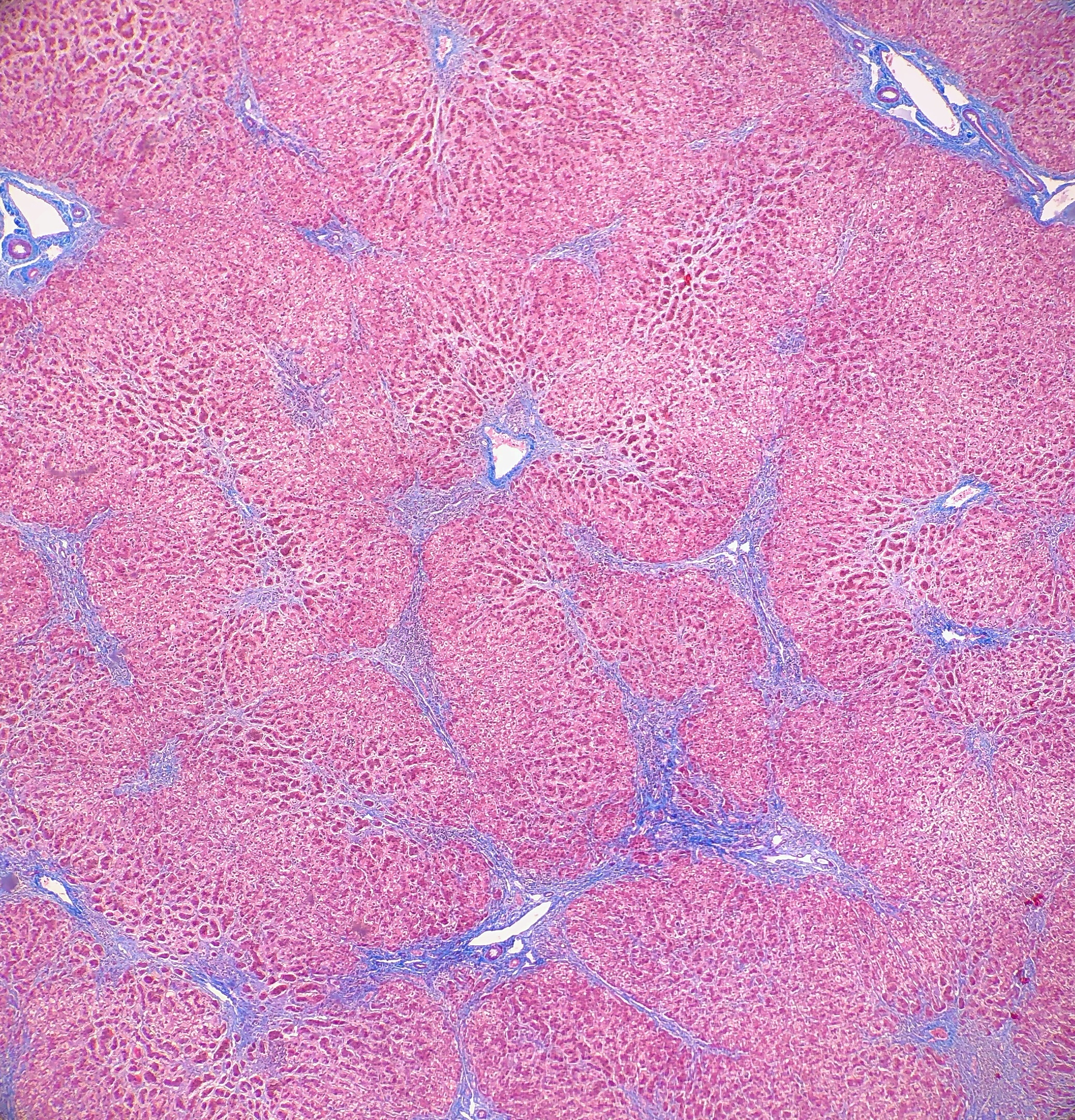
On
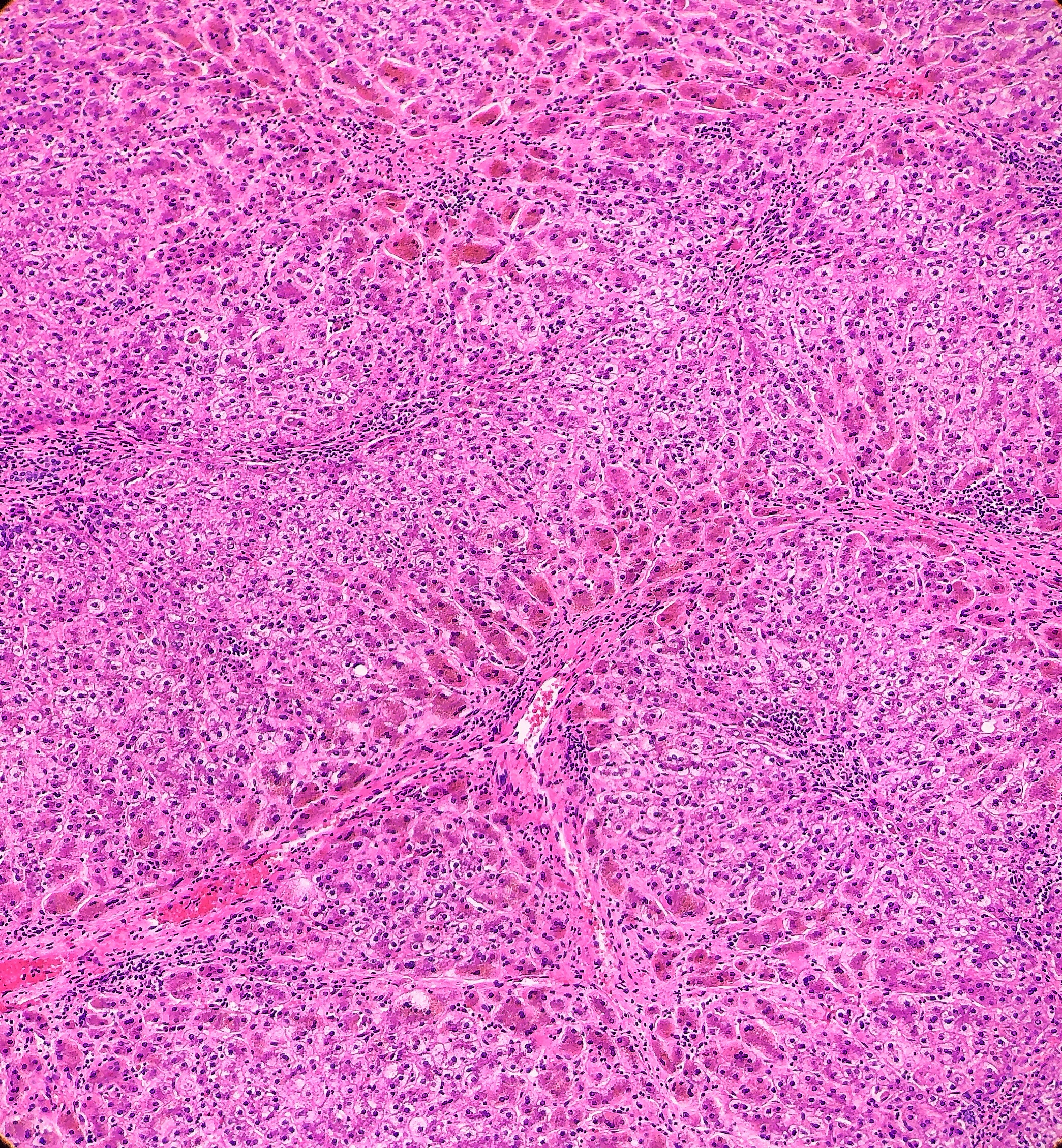
microscopy, the trichrome stain highlights the presence of portal and centrilobular
fibrosis, with focal bridging. However, regenerative nodule formation is not
evident. The portal tracts contain sparse mononuclear cell infiltrates. Significant
bile ductular proliferation is also evident, as confirmed by a CK7 immunostain.
However, the native bile ducts appear unremarkable. There is also considerable
hepatocellular and canalicular cholestasis in the centrilobular regions. Occasional
multinucleated hepatocytes are also seen within the centrolobular zones. No
steatosis is evident.
This
constellation of histologic features is consistent with the clinical history of
progressive familial intrahepatic cholestasis, type II.
Discussion
Progressive familial intrahepatic cholestasis
(PFIC) is a group of autosomal recessive disorders that affects bile formation
and results in cholestasis of the liver, usually beginning in infancy and
childhood. There are three types of PFIC, each related to a mutation in the
liver transport system genes that are involved in bile formation. PFIC type 1
(PFIC1), which is also referred to as Byler disease, is due to impaired bile
salt secretion related to a ATP8B1 gene that encodes the FIC1 protein. PFIC
type 2 (PFIC2), which is referred to as Byler syndrome, is due to
impaired bile salt secretion (similar to type 1), but is related to the ABCB11
gene that encodes the bile salt export pump, or BSEP. PFIC type 3 (PFIC3) is
due to impaired biliary phospholipid secretion that is related to a defect in
the ABCB4 gene that encodes the multi-drug resistant 3 protein, or MDR3.
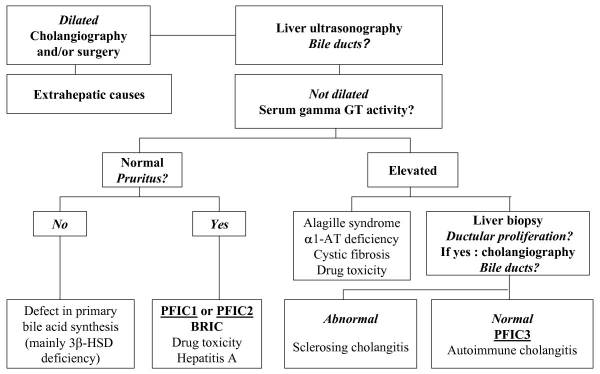
PFIC is suspected to be the cause of cholestasis
in 10-15% of children, and is also the underlying cause of liver transplants in
10-15% of children. The exact prevalence remains unknown, but is estimated to
be between 1 in every 50,000-100,000 births. PFIC1 and PFIC2 account for 2/3 of
all PFIC cases, with PFIC3 making up the other 1/3. PFIC is present worldwide,
and there does not appear to be a gender predilection.
The main clinical manifestation in all forms of PFIC, hence the
name, is cholestasis, and will usually appear in the first few months of life
with PFIC1 and PFIC2. Recurring episodes of jaundice are also present in PFIC1,
whereas permanent jaundice and a rapid evolution to liver failure are
characteristic of PFIC2. In PFIC3, cholestasis is noted within the first year
of life in 1/3 of all cases, but rarely will be present in the neonatal period.
PFIC3 can also present later in infancy, childhood or even early adulthood,
with gastrointestinal bleeding due to portal hypertension and cirrhosis being
the main symptoms that the patient would present with. Pruritus is severe in
PFIC 1 and 2, but has a more mild presentation in PFIC3. There have been multiple
cases reported of hepatocellular carcinoma that are associated with PFIC2, but
there so far have not been any cases of hepatocellular carcinoma reported that
are associated with PFIC3. Other signs and symptoms that may be present in
PFIC1 include short stature, deafness, diarrhea, pancreatitis and liver
steatosis. When examining clinical laboratory results, patients with PFIC1 and
PFIC 2 will have normal serum gamma-glutamyltransferase (GGT) levels, but
patients with PFIC3 will have elevated GGT levels. PFIC1 and PFIC2 can be
differentiated from each other by the higher transaminase and alpha-fetoprotein
levels that are found in PFIC2. When analyzing the biliary bile salt
concentrations, PFIC1 will have mildly decreased levels (3-8 mM), PFIC2 will
have drastically decreased levels (<1 mM), and PFIC3 will have normal
levels. In addition, the biliary bile salt:phospholipid ratio and the
cholesterol:phospholipid ratio will be approximately 5 times higher in PFIC3
than in normal bile, due to the biliary phospholipid levels being dramatically
decreased (normal phospholipid range = 19-24%, PFIC phospholipid range =
1-15%).
Histologically, PFIC1 and PFIC 2 will have canalicular
cholestasis, an absence of true ductular proliferation, and periportal biliary
metaplasia of the hepatocytes. In PFIC2, these manifestations are much more
worrisome with more marked lobular and portal fibrosis, and inflammation, as
well as having much more pronounced necrosis and giant cell transformation (Images
3 and 4). PFIC3 will show portal fibrosis and true ductal proliferation, with a
mixed inflammatory infiltrate. In addition, cholestasis can be present in the
lobule and in some of the ductules that contain bile plugs. Cytokeratin
staining can help confirm the ductular proliferation within the portal tract.
Mild or absent canalicular staining with BSEP and MDR3 antibodies will help to
diagnose PFIC2 and PFIC3, respectively.
A diagnosis of PFIC is based on the clinical manifestations, liver ultrasonography, cholangiography and liver histology, as well as on specific tests for excluding other causes of childhood cholestasis (such as biliary atresia, Alagille syndrome, cystic fibrosis and alpha-1 antitrypsine deficiency). Ultrasonography of the liver will be normal with the exception of a possible dilated gallbladder. At the time of the liver biopsy, a portion of tissue can be submitted for electron microscopy, which in the case of PFIC, can show canalicular dilatation, microvilli loss, abnormal mitochondrial internal structures, and varying intra-canalicular accumulations of bile. PFIC1 will have coarsely, granular bile on electron microscopy, whereas PFIC2 will have a more amorphous appearance. If biliary obstruction is noted on the liver biopsy, a cholangiography will need to be performed to exclude sclerosing cholangitis. If a normal biliary tree is observed, as in PFIC, bile can be collected for biliary bile salt analysis (which was discussed earlier in the laboratory results section). Differentiating between PFIC1, PFIC2 and PFIC3 can be quite troublesome, but luckily Davit-Spraul, Gonzales, Baussan and Jacquemin proposed a fantastic schematic for the clinical diagnosis of PFIC, which is presented as Figure 1.
Figure 1. Schematic proposed for the clinical diagnosis of progressive familial intrahepatic cholestasis
Ursodeoxycholic acid (UDCA) therapy should be considered in all patients with PFIC to prevent liver damage and provide relief from pruritus. Rifampicin and Cholestyramine can help in cases of PFIC3, but have been found to provide no improvement in PFIC1 or PFIC2. In some PFIC1 or PFIC2 patients, biliary diversion can also relieve pruritus and slow disease progression. The total caloric intake should be around 125% of the recommended daily allowance. Dietary fats should come in the form of medium chain triglycerides, and care should be taken to check the patient’s vitamin levels to look for signs of vitamin deficiency. Patients with PFIC2 should be monitored for hepatocellular carcinoma, beginning from the first year of life. Ultimately, most PFIC patients develop fibrosis and end-stage liver disease before adulthood, and are candidates for liver transplantation. Diarrhea, steatosis and short stature may not improve after liver transplantation, and could become aggravated from the procedure. Hepatocyte transplantation, gene therapy or specific targeted pharmacotherapy are possible alternative therapies for PFIC, but will require more research and studies to determine whether they are viable options.
References
Davit-Spraul A, Gonzales E, Baussan
C, Jacquemin E. Progressive familial intrahepatic cholestasis. Orphanet
J Rare Dis. 2009;4(1). doi:10.1186/1750-1172-4-1
Evason K, Bove KE, Finegold MJ, et al. Morphologic findings
in progressive familial intrahepatic cholestasis 2 (PFIC2): correlation with
genetic and immunohistochemical studies. Am J Surg Pathol.
2011;35(5):687–696. doi:10.1097/PAS.0b013e318212ec87
-Cory Nash is a board certified Pathologists’ Assistant, specializing in surgical and gross pathology. He currently works as a Pathologists’ Assistant at the University of Chicago Medical Center. His job involves the macroscopic examination, dissection and tissue submission of surgical specimens, ranging from biopsies to multi-organ resections. Cory has a special interest in head and neck pathology, as well as bone and soft tissue pathology. Cory can be followed on twitter at @iplaywithorgans.