Case History
The patient is a 72 year old
woman who presented to her physician’s office with postprandial pain and
unintentional weight loss. A CT scan was performed that showed no obvious
abnormality or cause for the patient’s abdominal pain. The patient subsequently
underwent an EGD and EUS which revealed enlarged gastric folds
without hemorrhage. In addition, there was wall
thickening seen in the body of the stomach within the luminal interface, superficial mucosa, deep mucosa
and submucosa consistent with possible
gastritis versus an infiltrative process. The remainder of the EGD and EUS was
grossly unremarkable. These findings
were concerning for possible linitis plastica. Pathology on the samples taken from the EGD were consistent
with poorly differentiated adenocarcinoma
that was invasive in both the gastric fundus and gastric body. The patient was initially taken to the operating room for
a staging laparoscopy to ensure that there was no metastatic disease before
beginning a preoperative chemotherapy regimen. The staging laparoscopy revealed
a thickened gastric wall from the fundus to the antrum, consistent with linitis
plastica, and no obvious evidence of metastatic disease. The patient then
underwent peritoneal washings which showed no evidence of positive cytology. Based
on these findings, the patient was started on a chemotherapy regimen of
epirubicin, cisplatin and fluorouracil (5-FU), which she tolerated well. The
patient was then taken to the operating room for a total gastrectomy procedure
with Roux-en-Y esophagojejunostomy.
Diagnosis
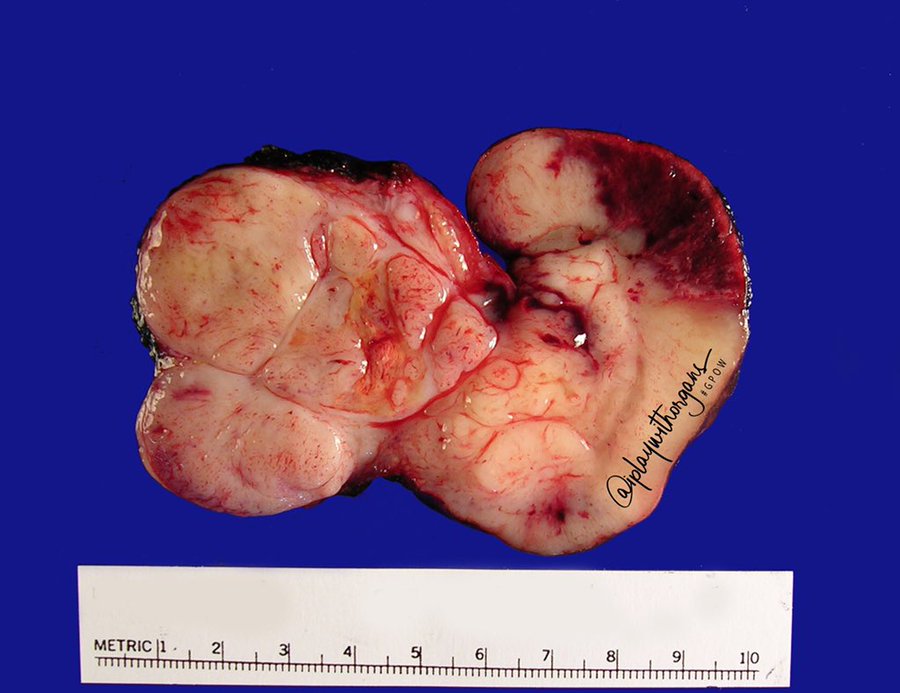
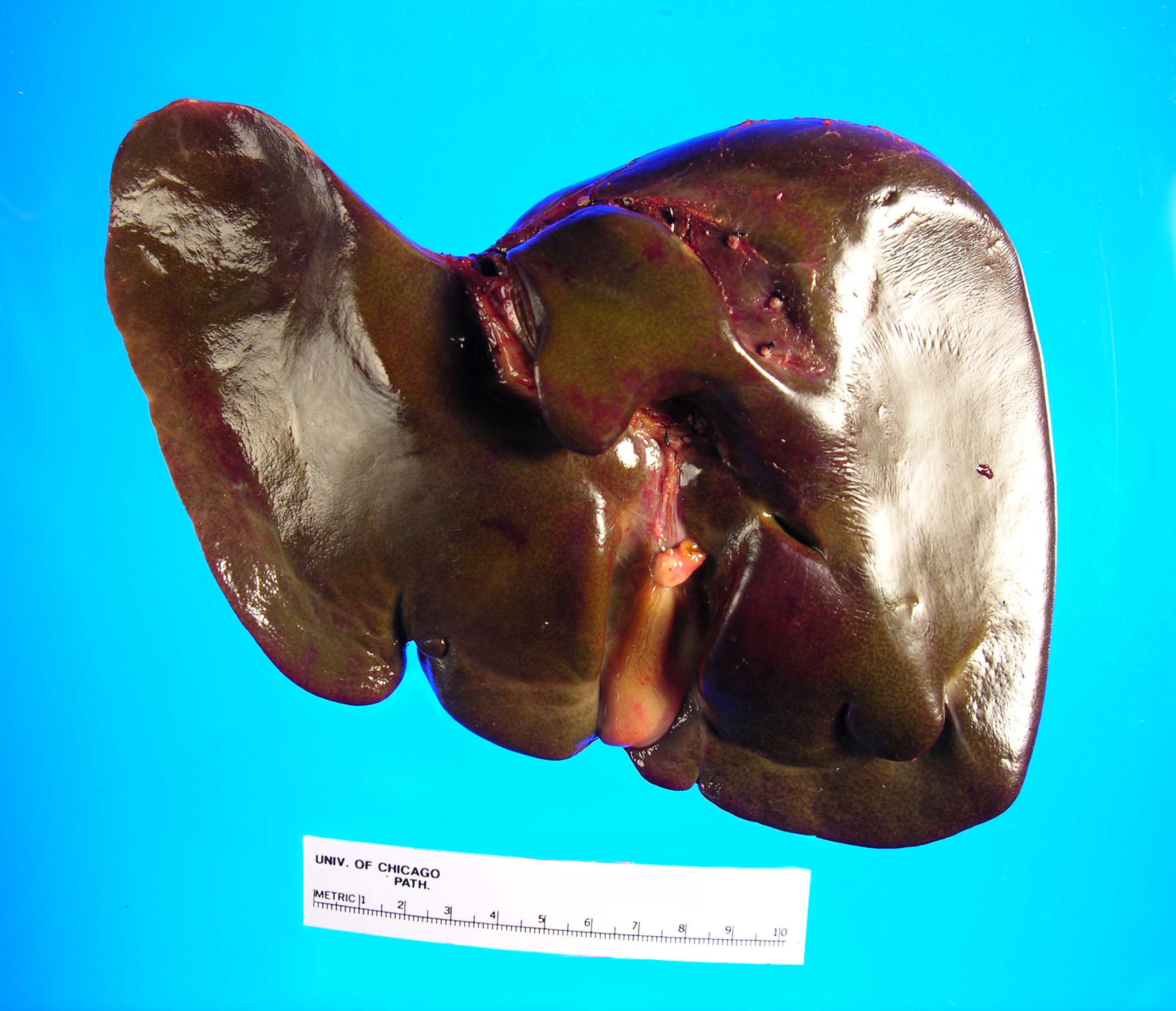
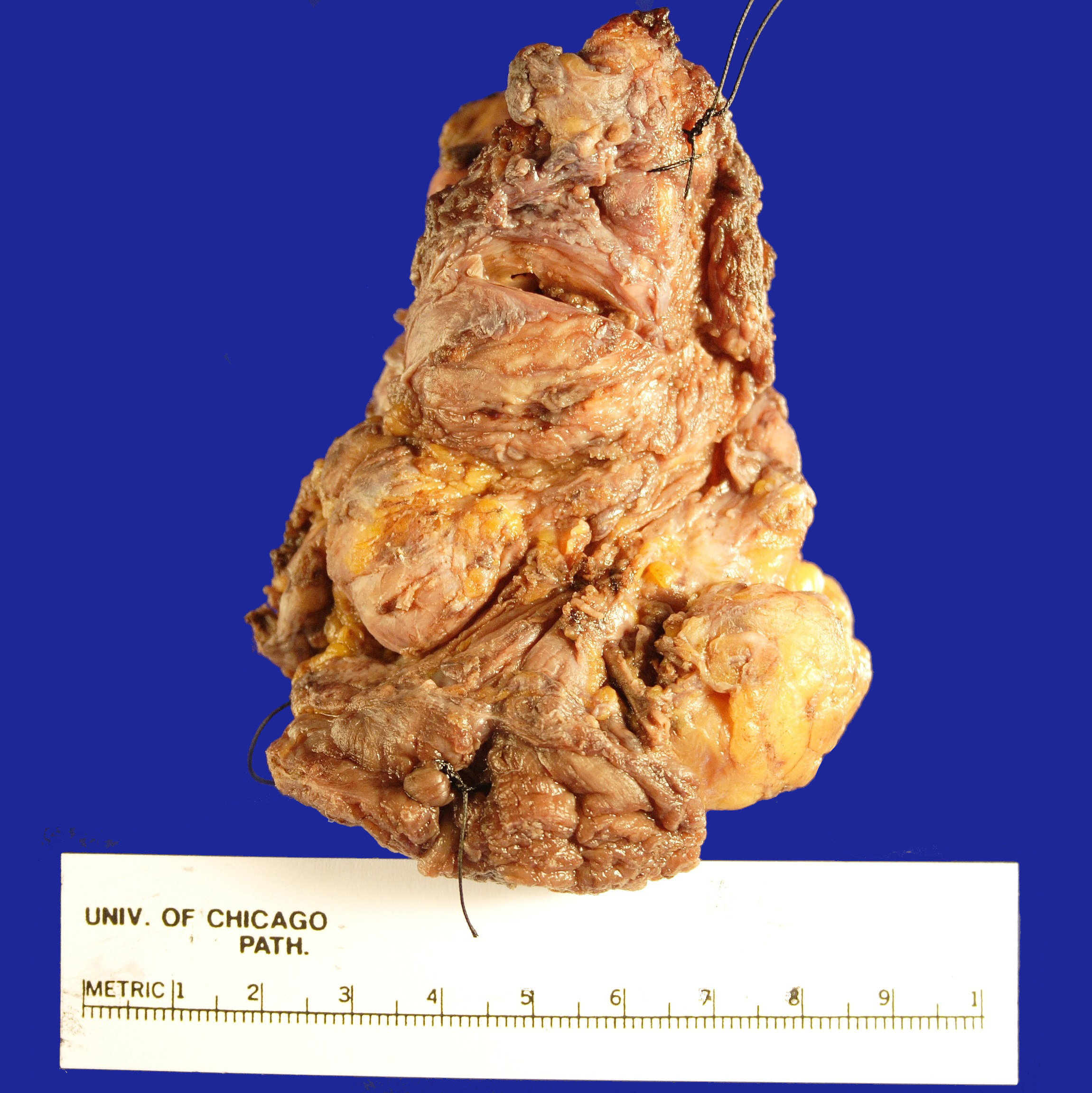
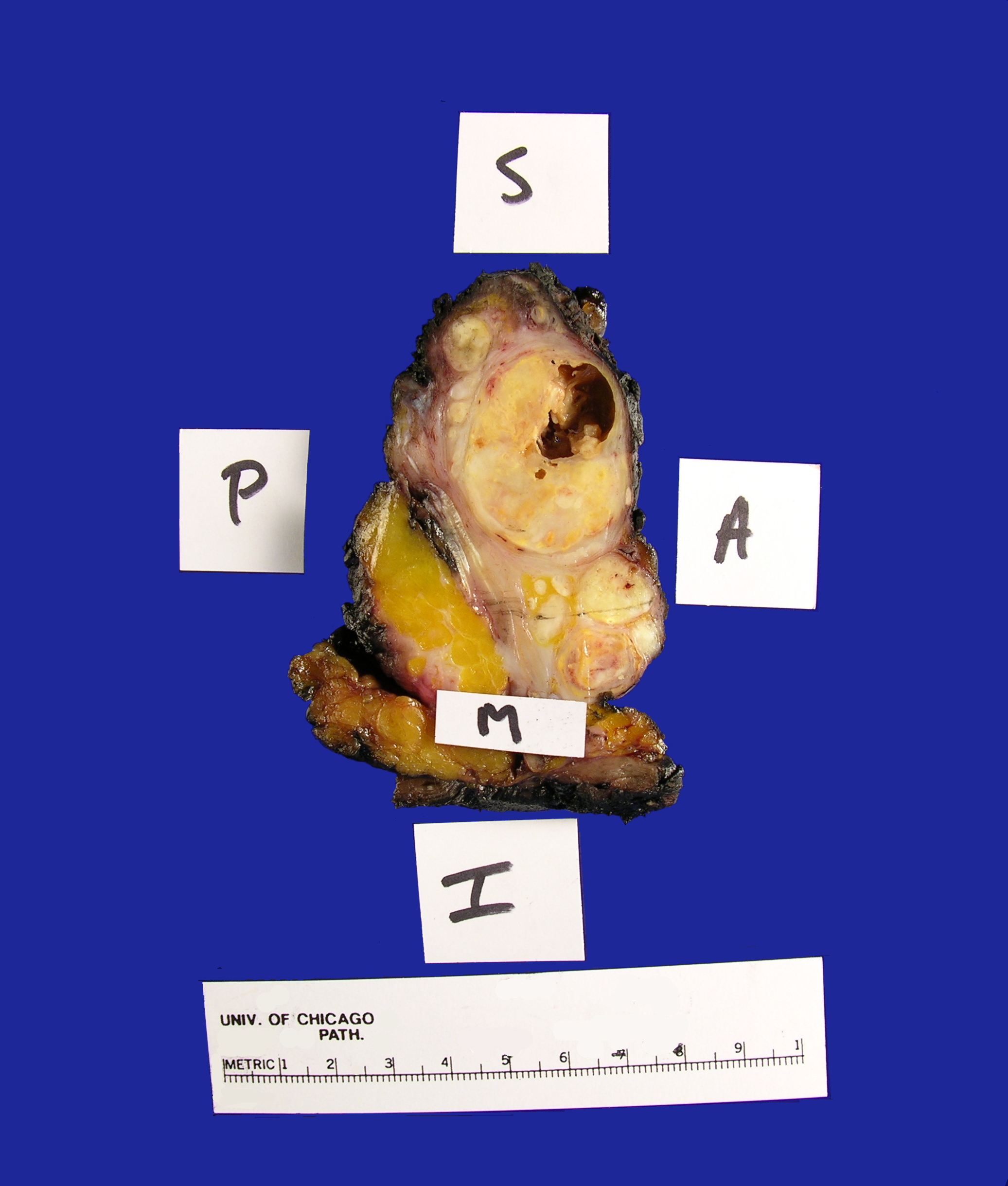
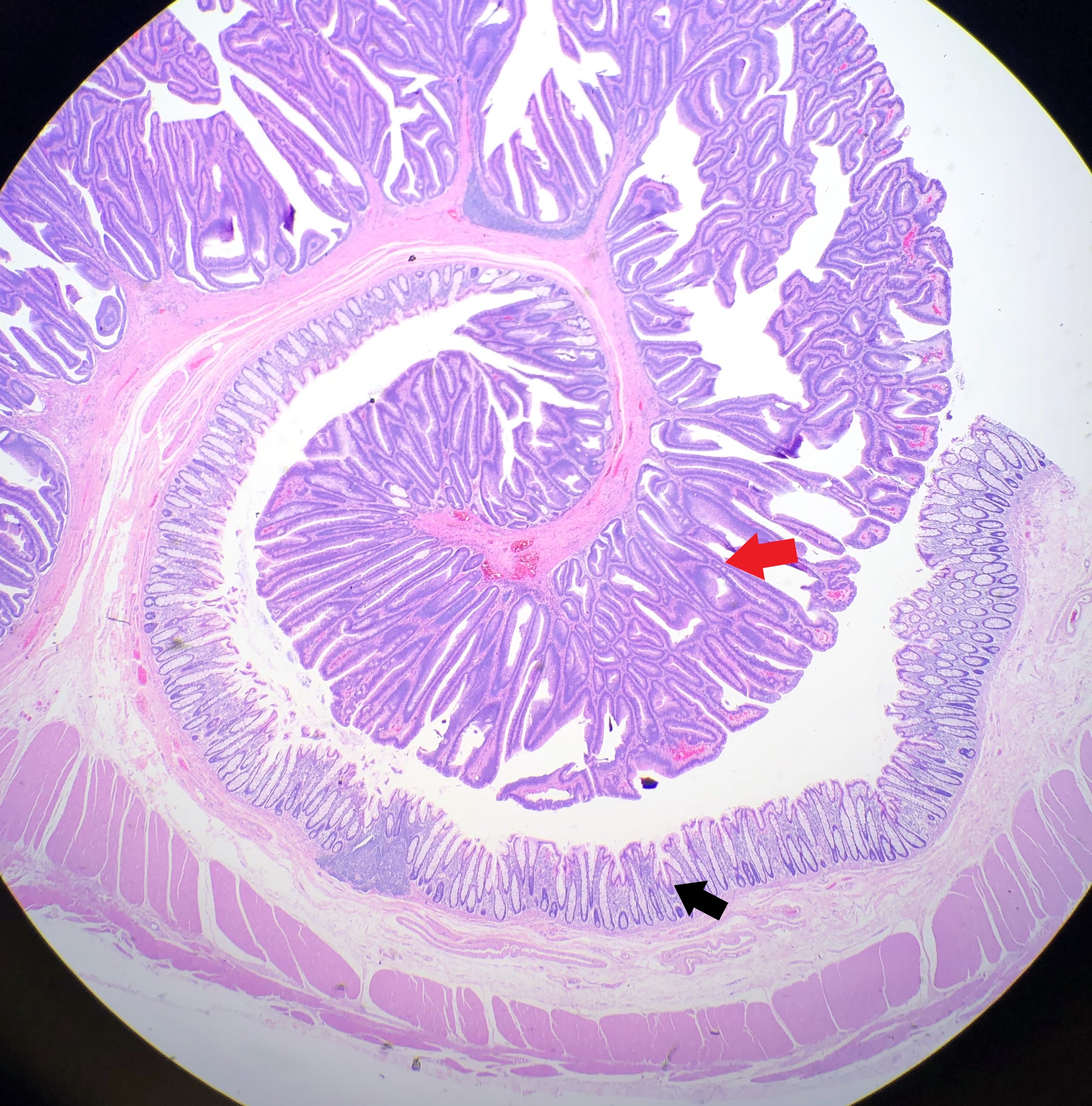
Received fresh for intraoperative consultation is a total gastrectomy specimen with a black stitch designating the proximal side. It was requested by the surgical team to have the proximal esophageal margin frozen to ensure that esophageal tissue was indeed present, as well as to exclude the presence of any carcinoma. The proximal margin was negative for carcinoma with squamous mucosa present. The stomach measures 17.0 cm in length with an internal circumference ranging from 14.7 cm proximally to 9.0 cm distally. There is a 1.0 cm long portion of attached duodenum with an internal circumference of 5.8 cm. The serosal surface of the stomach is glistening, pink-tan and smooth with a scant amount of attached yellow, lobulated adipose tissue and omentum along the length of one entire edge measuring 26.0 x 13.0 x 1.0 cm. The stomach is opened to reveal glistening, tan mucosa with irregular rugal folds which are diffusely nodular, predominantly in the body of the stomach. There is a 6.5 x 5.0 cm are of flattened mucosa in the pyloric region (Image 1). The wall thickness measures 0.5 cm throughout. There are no grossly identifiable masses or nodules. Gross images are taken and the serosal surface is inked entirely in black. The adipose tissue is examined for candidate lymph nodes. Representative sections are submitted as follows:
B1 FS: Frozen section remnants
B2-B6: multiple representative sections from the cardia
B7-B10: multiple representative sections from the body
B11-B12: multiple representative sections from the pylorus
B13: representative perpendicular section through the distal resection margin
B14: seven putative lymph nodes
B15: five putative lymph nodes
B16: three putative lymph nodes
B17: seven putative lymph nodes
B18: six putative lymph nodes
B19: three putative lymph nodes
B20: six putative lymph nodes
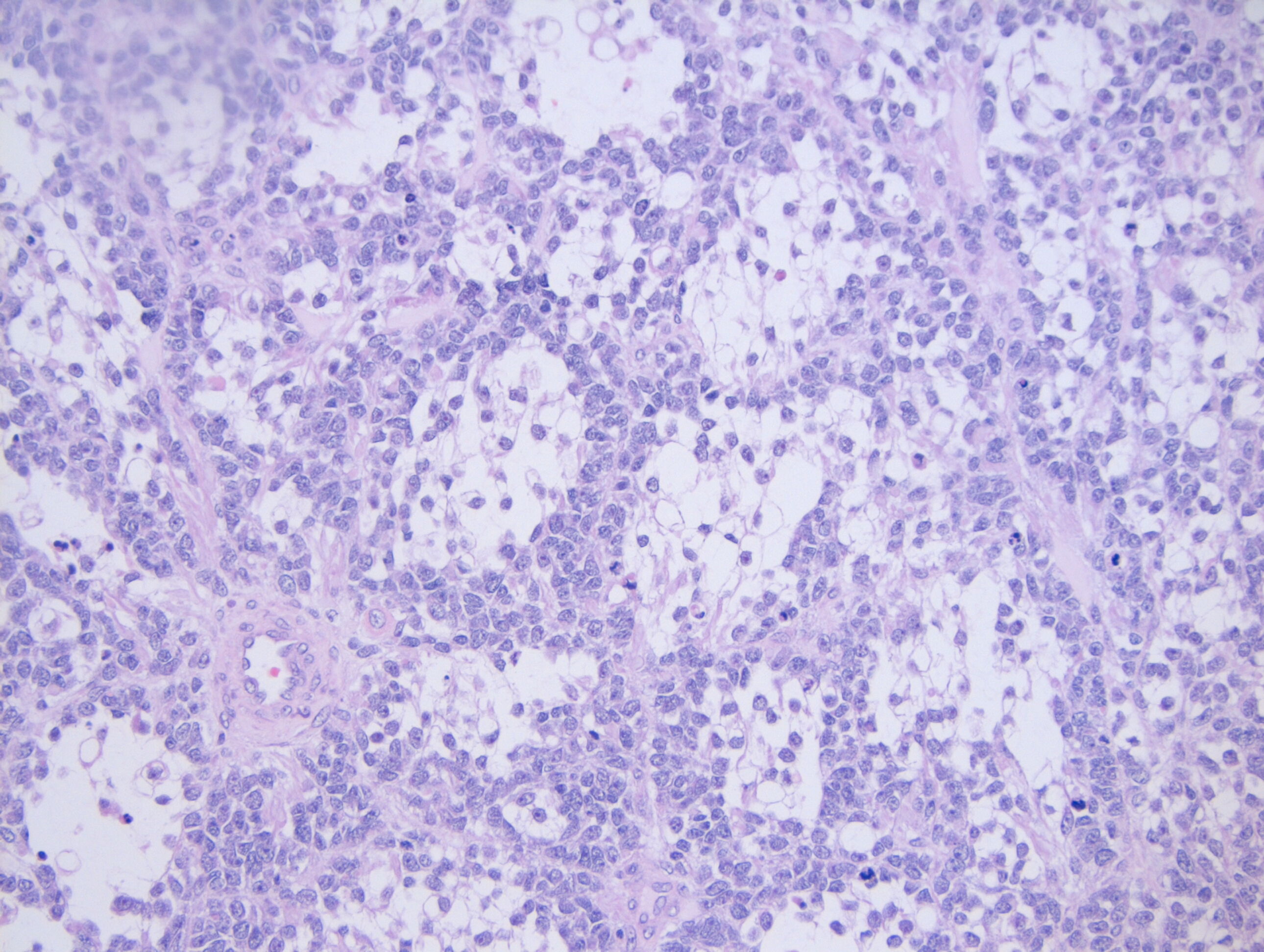
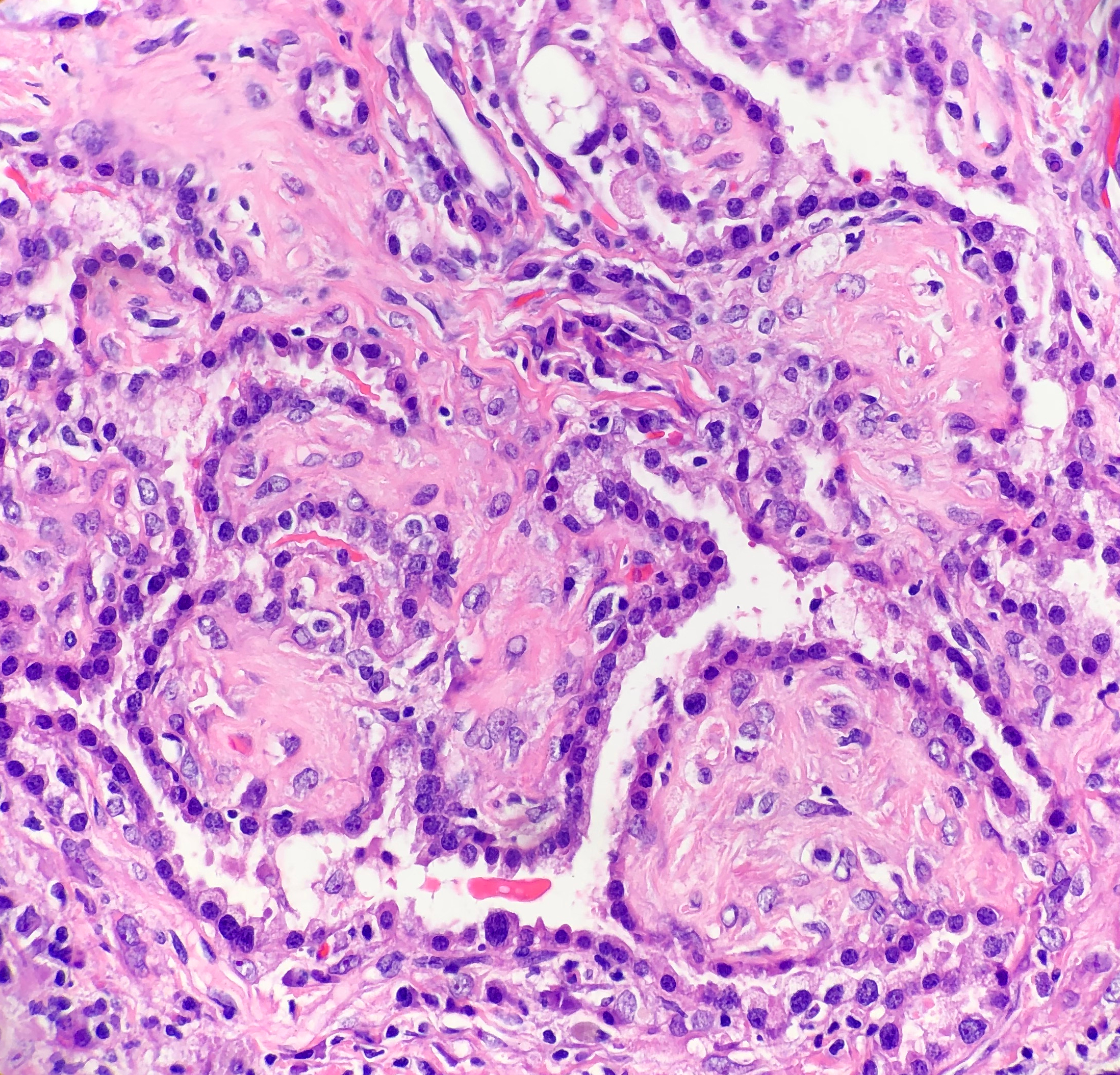
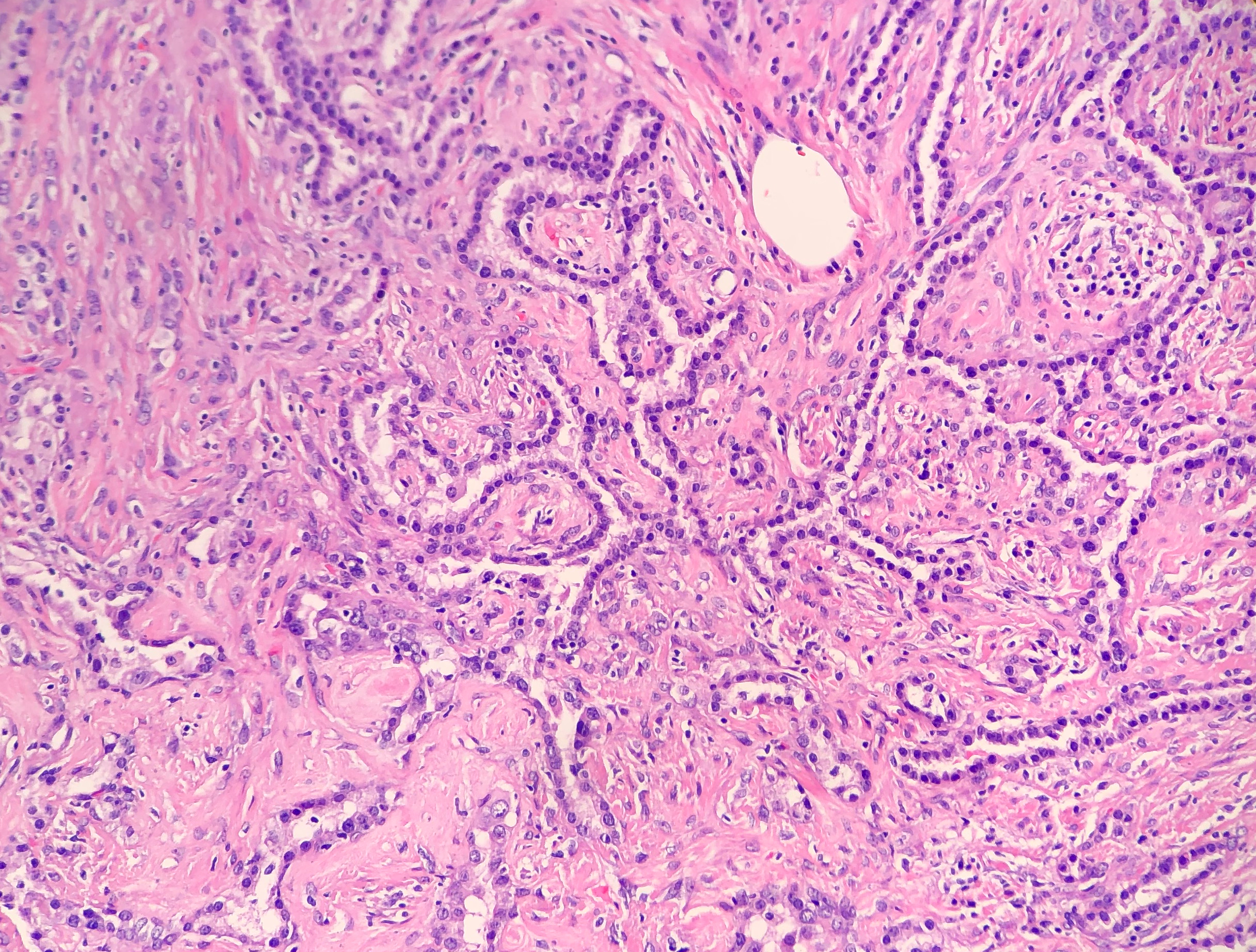
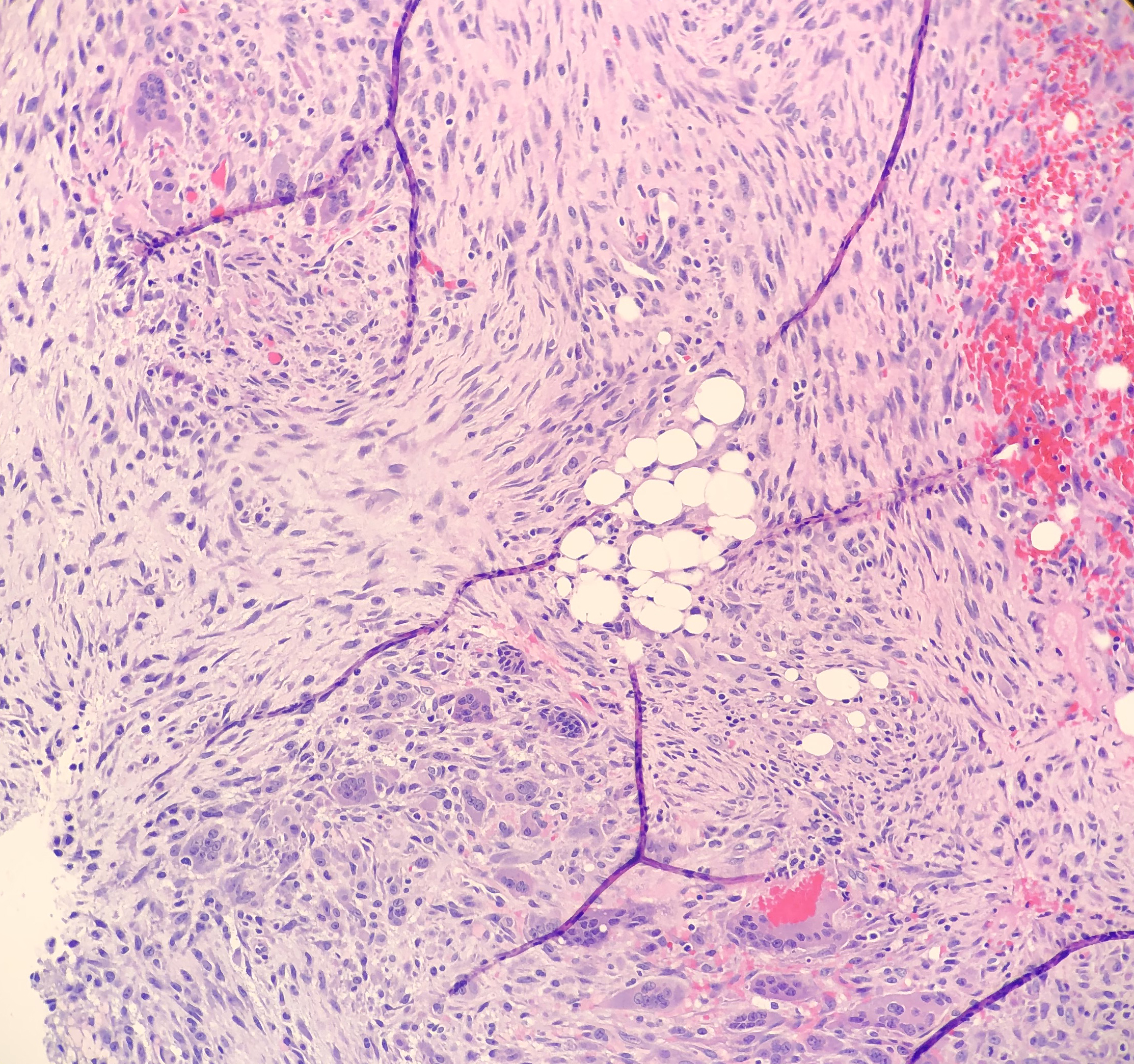
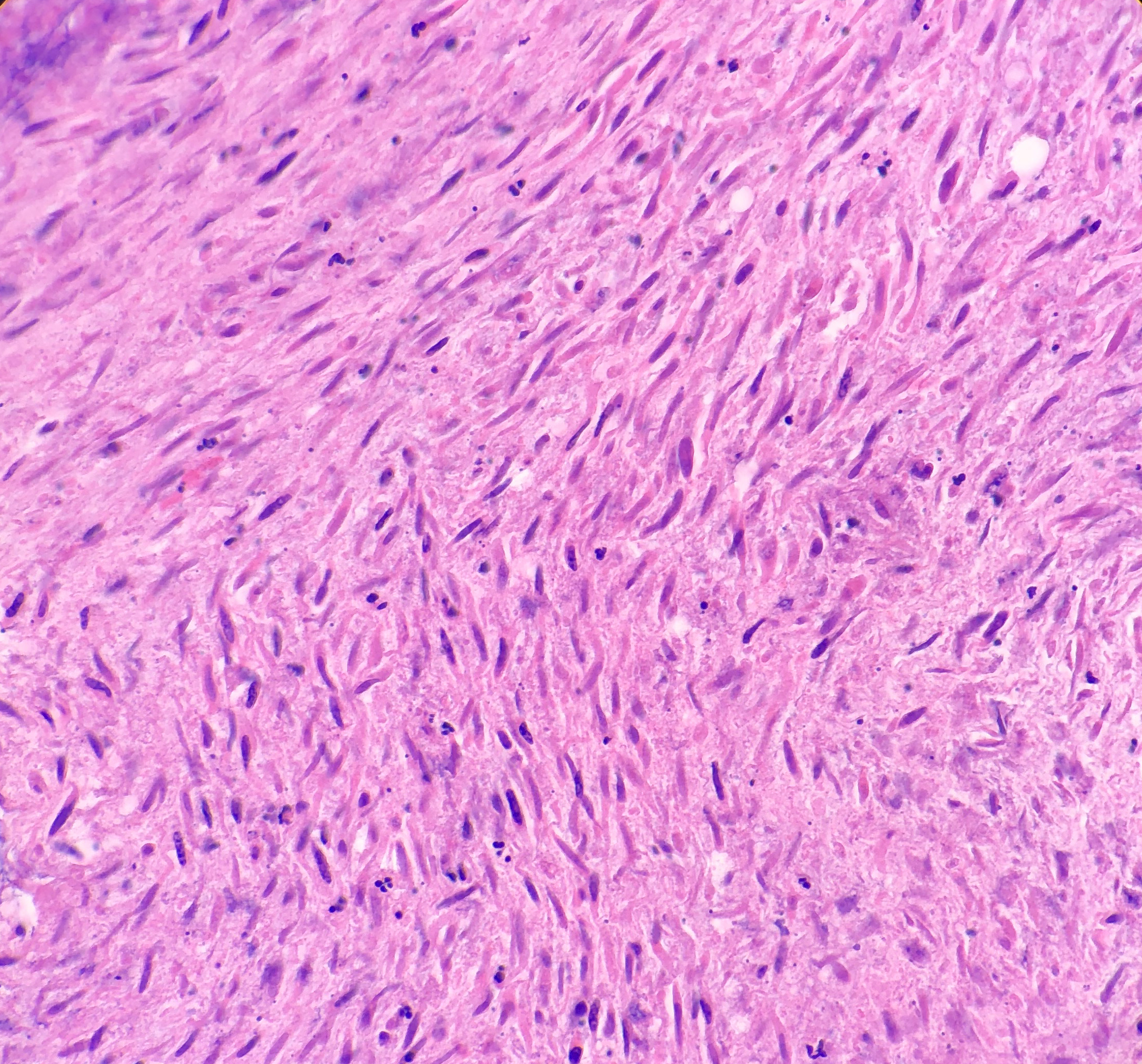
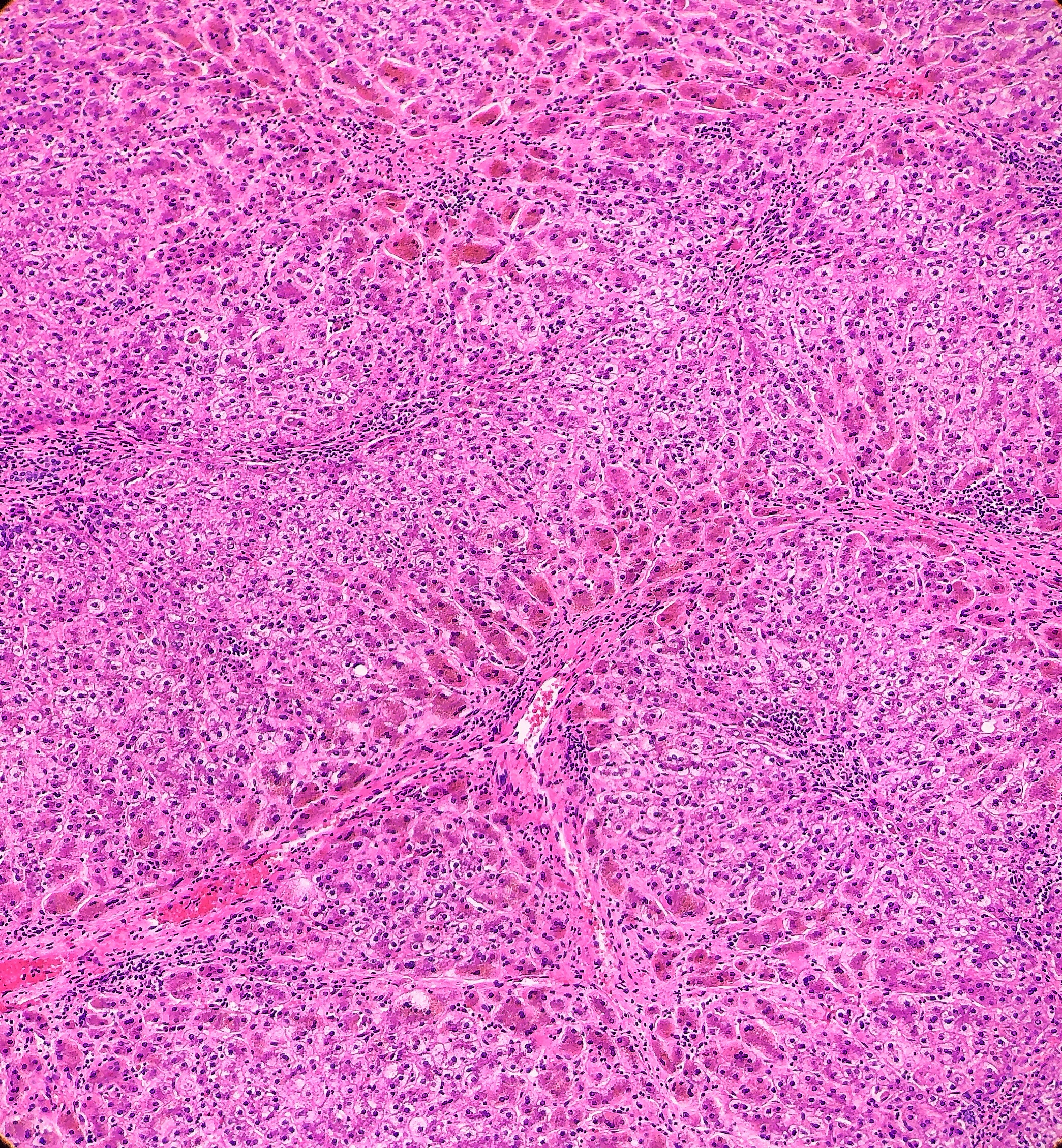
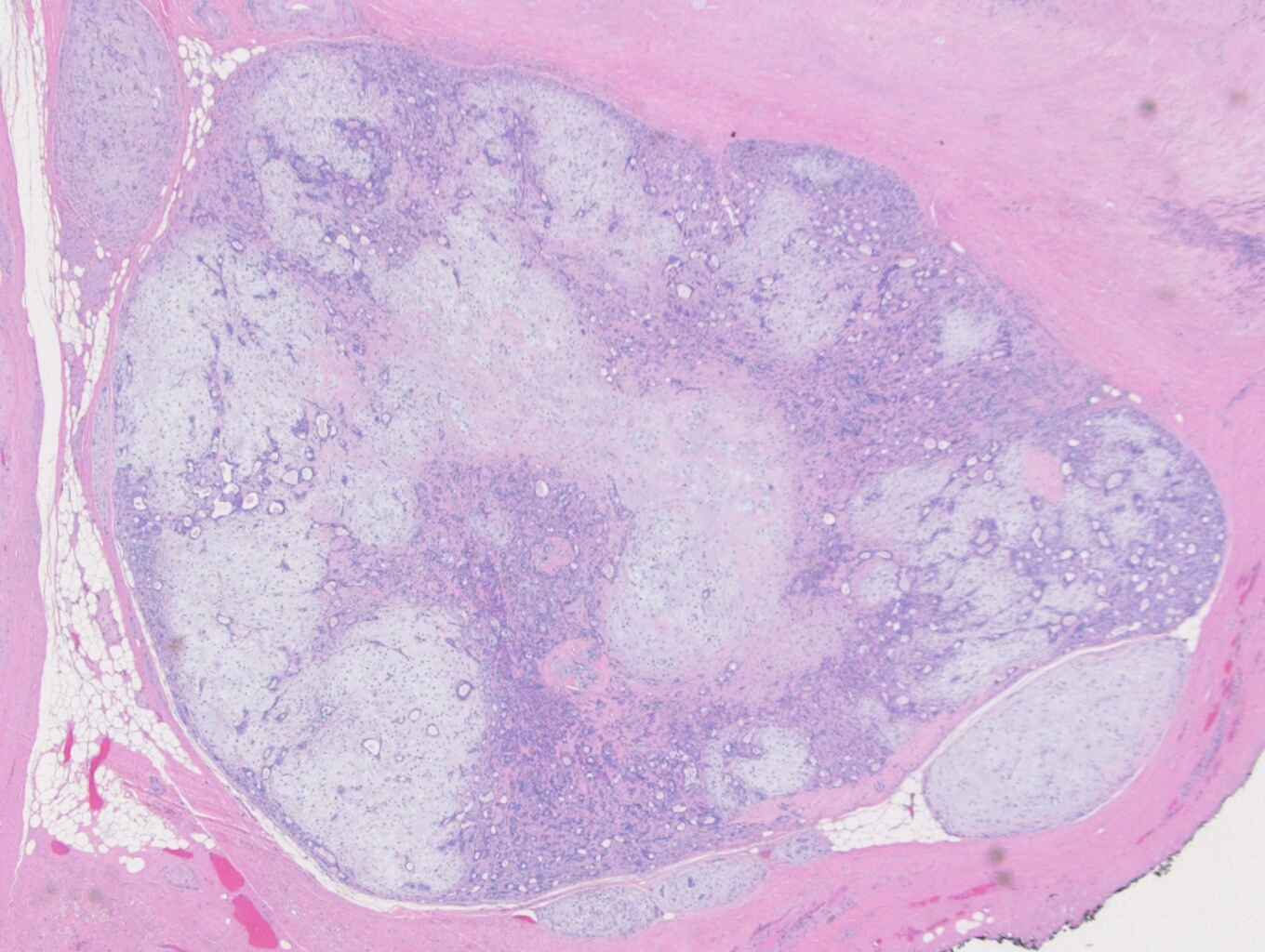
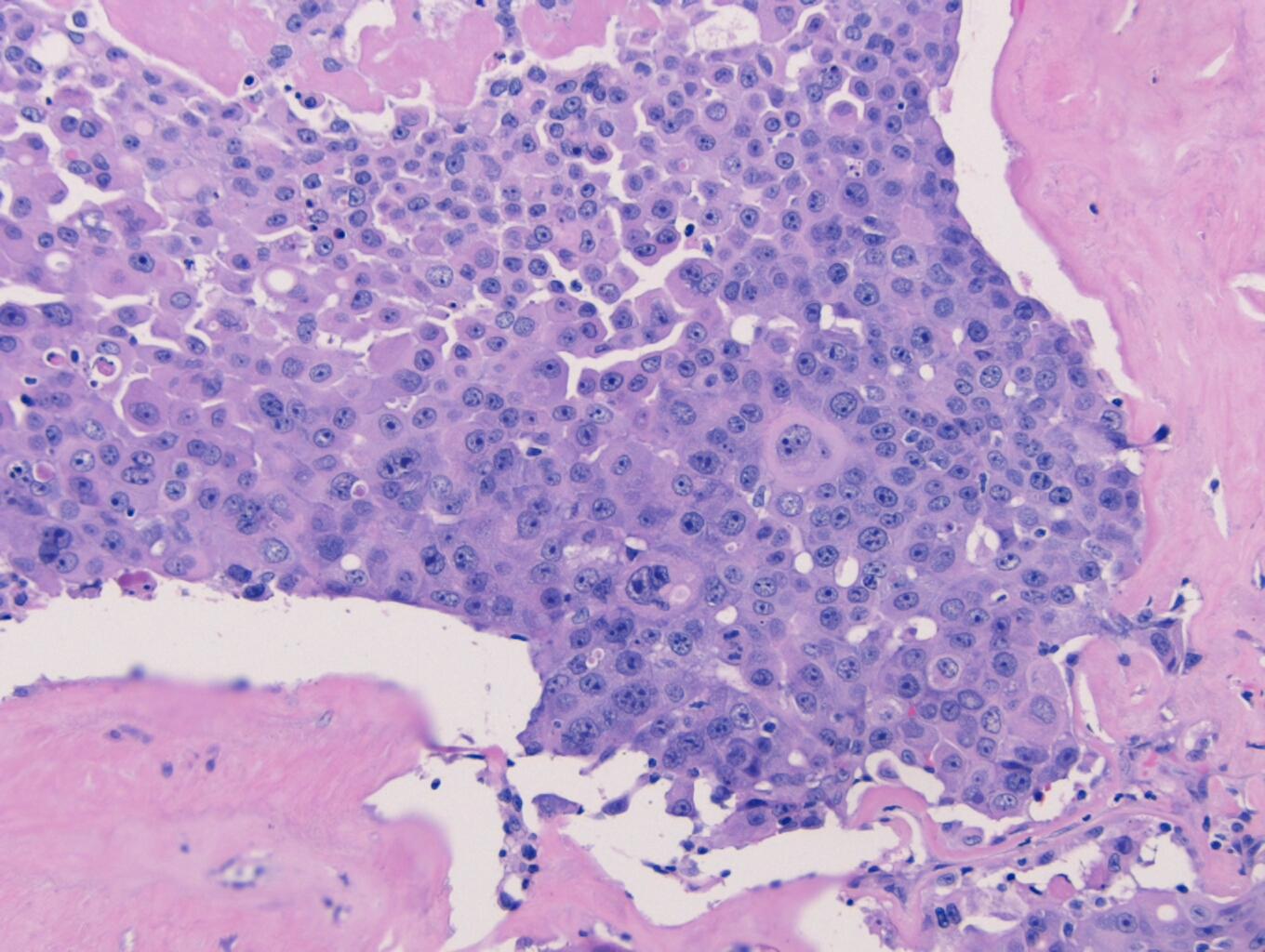
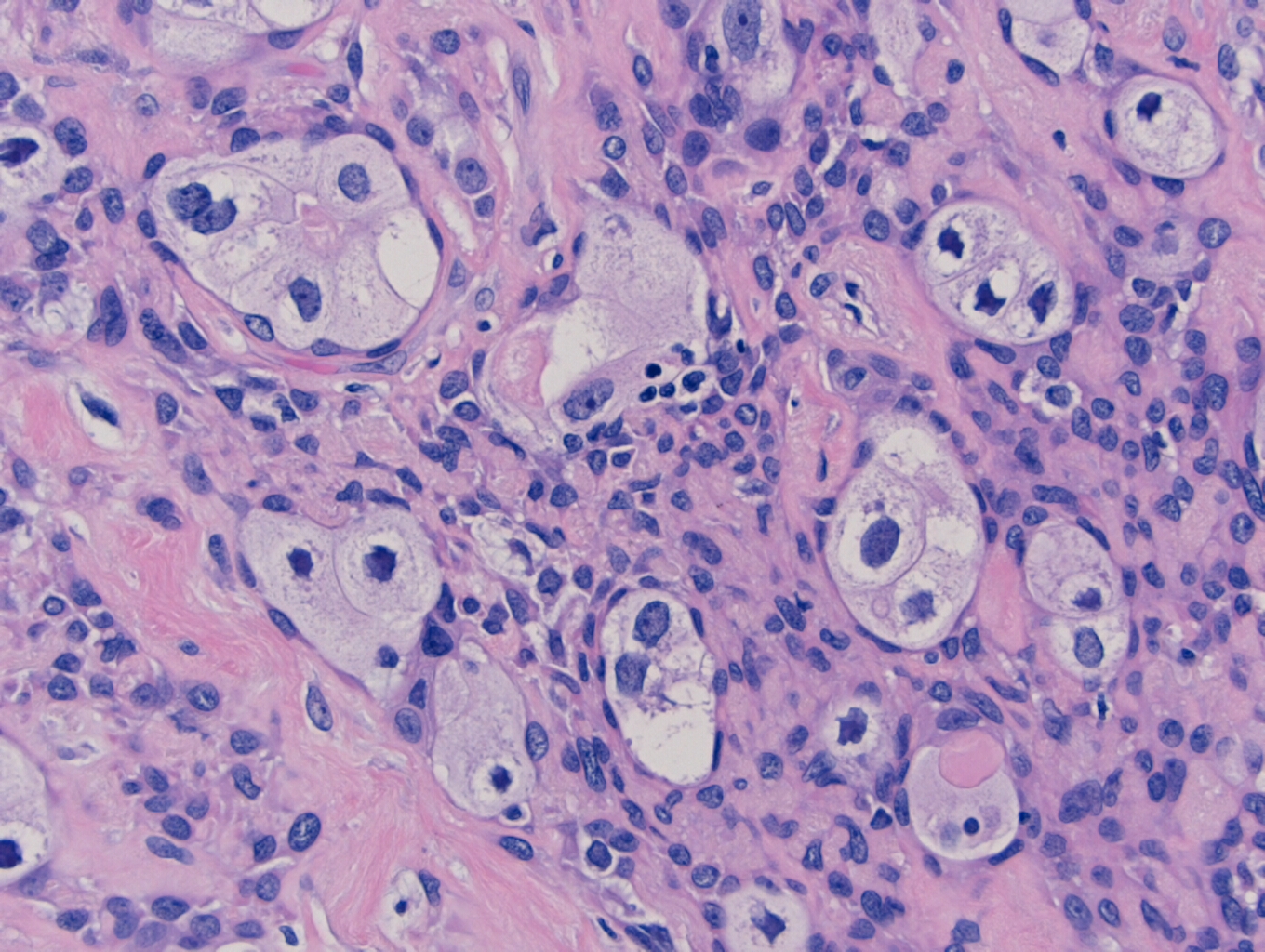
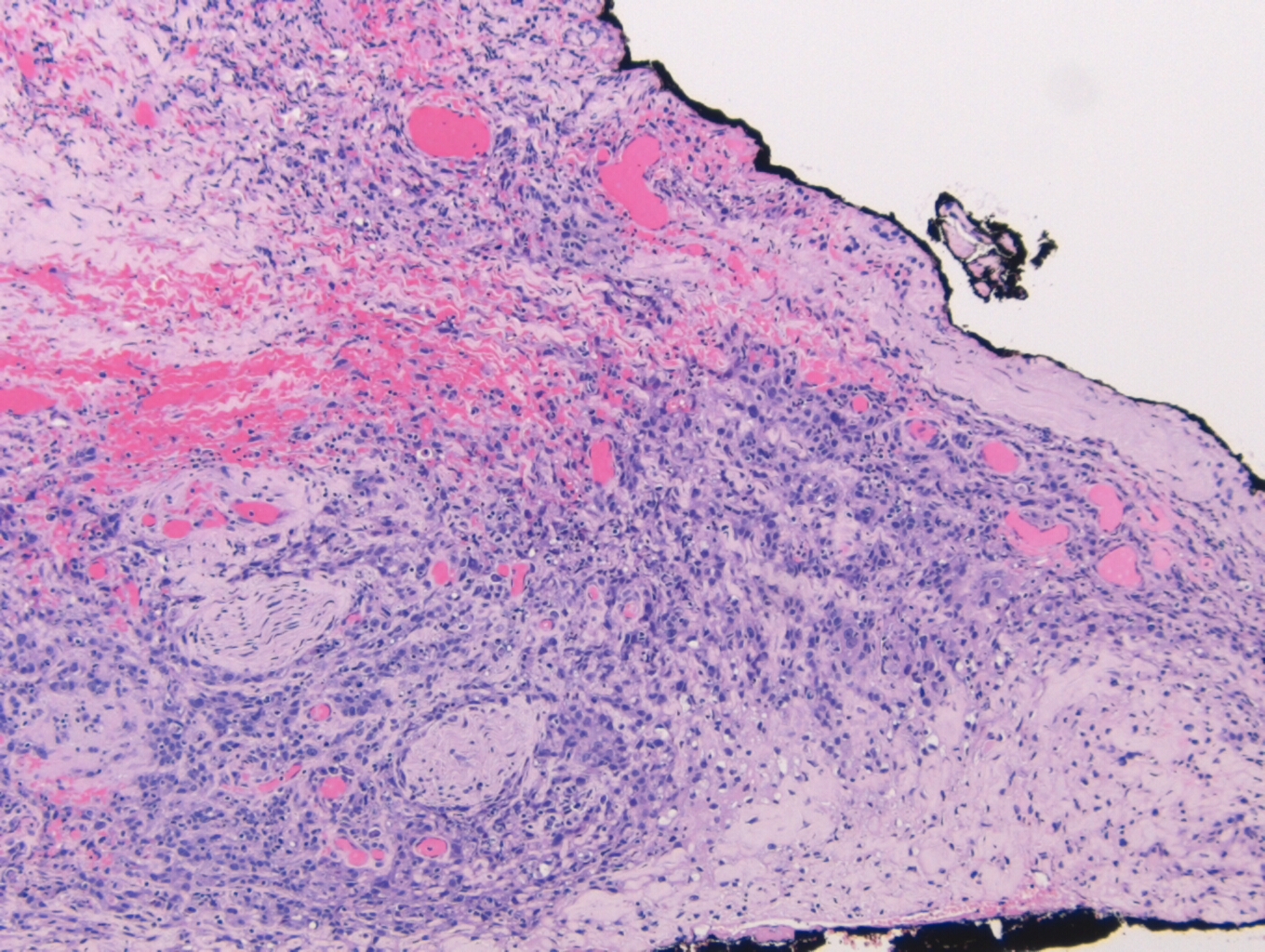
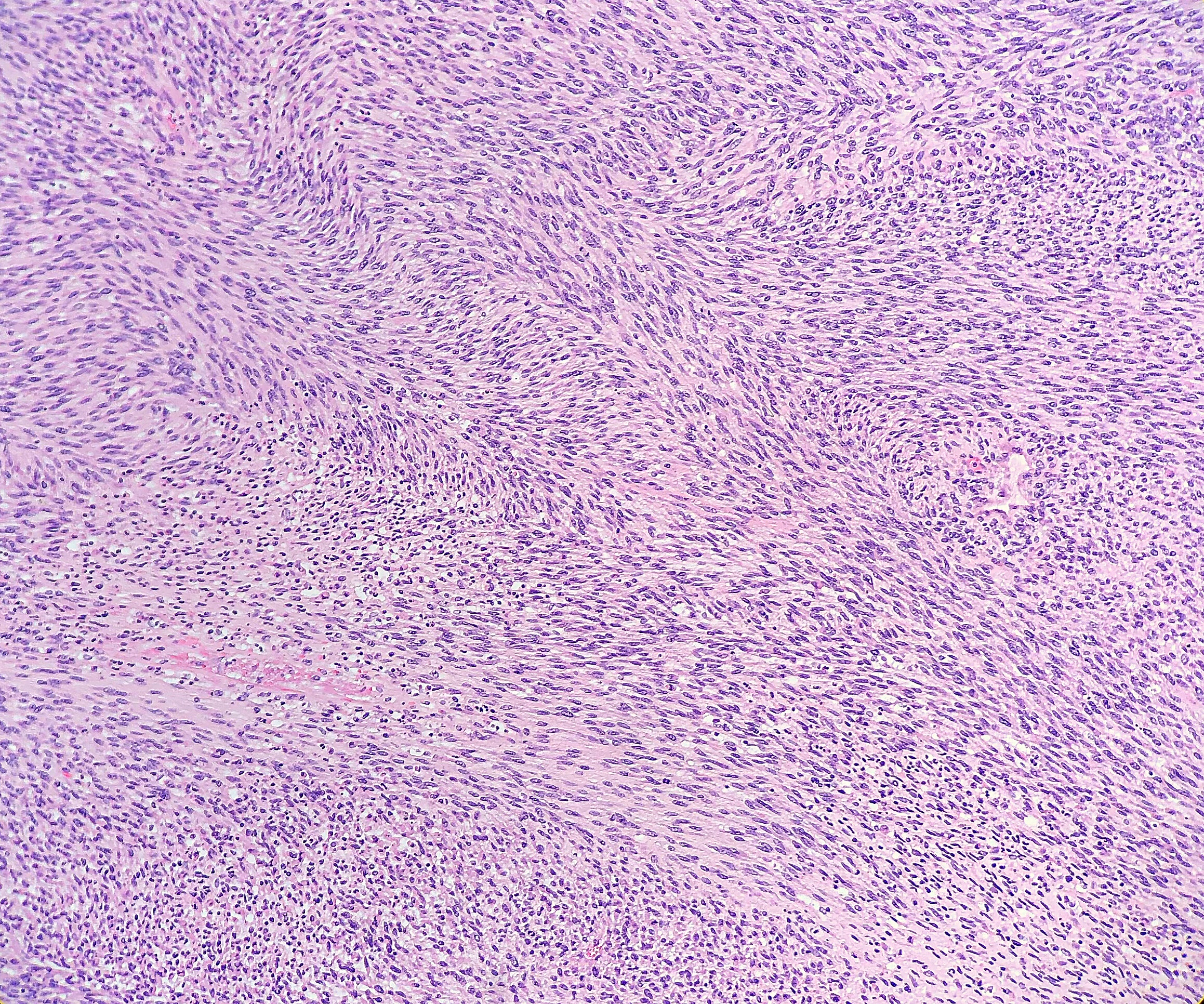
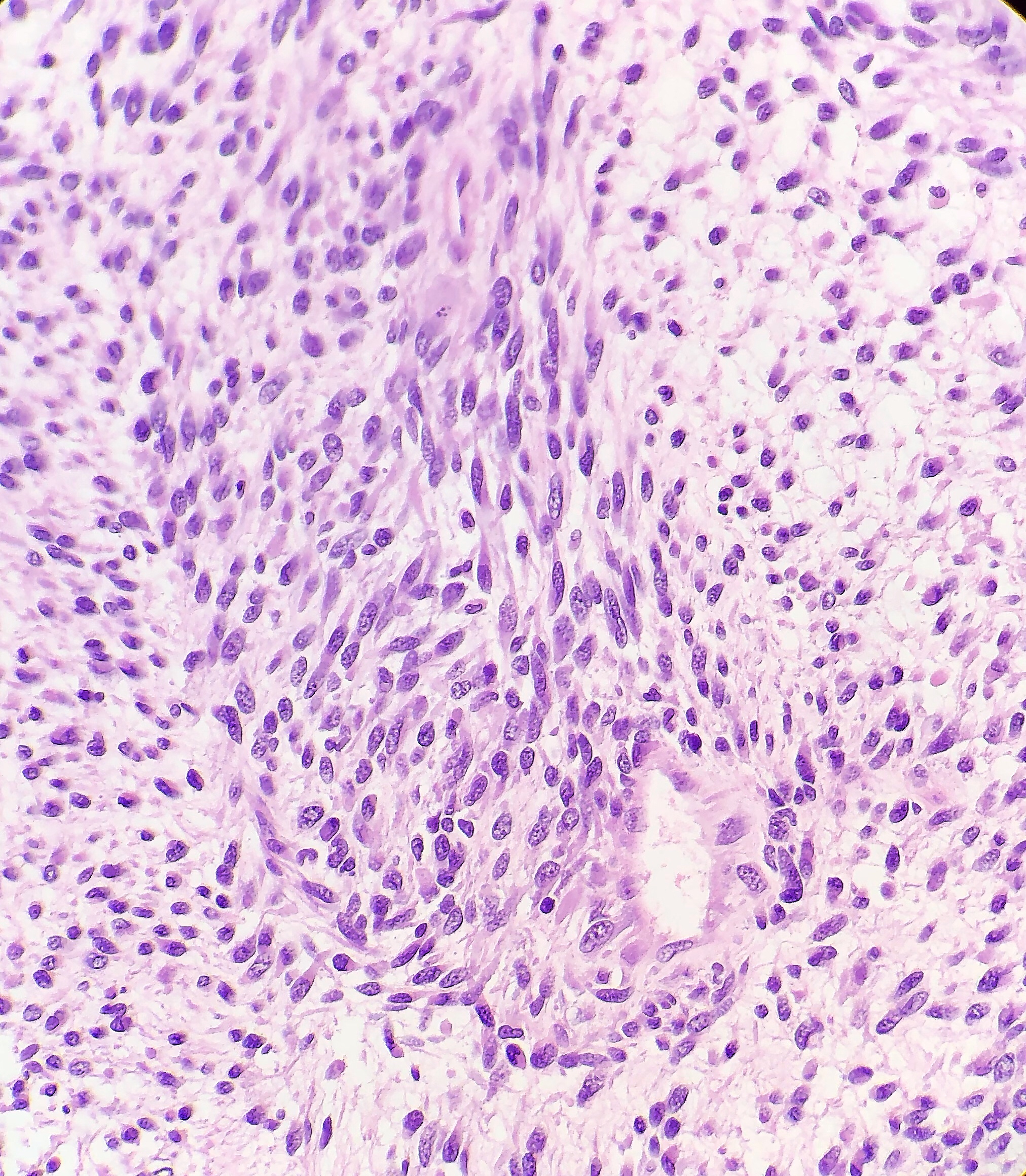
Histologically, the specimen consisted of diffuse, poorly differentiated, discohesive cells throughout all the layers of the stomach, penetrating into the serosa, with fibrosis, inflammation and signet ring cells present. In addition, angiolymphatic invasion was present. Based on the gross presentation and histologic appearance, the specimen was signed out as a diffuse gastric adenocarcinoma with a stage of T3.

Discussion
As of 2018, gastric cancer is the sixth most common cancer with approximately 1.03 million cases, and the third leading cause of cancer deaths worldwide, resulting in 783,000 deaths. Due to a better understanding of epidemiology, pathology, and molecular testing, as well as advances in new forms of treatments, the incidence and mortality in gastric cancer has been declining over the years. Of the gastric cancer types, rates of intestinal type carcinoma have been decreasing, however, the incidence of poorly cohesive gastric carcinoma (PCGC) and signet ring cell carcinoma (SRC) has increased. In order to accurately discuss PCGC, there must first be a discussion about the standardization of gastric cancer subtype definitions. Poorly cohesive, signet ring cell, and diffuse gastric carcinomas have commonly been used interchangeably. In 2010, the World Health Organization defined poorly cohesive gastric carcinoma as being composed of isolated or small groups of tumor cells. If there was a predominance of signet ring cells, then it would be termed a signet ring cell carcinoma. Mariette et al. proposed that a PCGC composed of 90% or more signet ring cells should be classified as SRC. The term “diffuse” corresponds to the same term “poorly cohesive”, and because of this, I will be using the term “poorly cohesive” solely going forward. In addition to this, the term “linitis plastica” would commonly be used interchangeably, but is best used as a term to describe the macroscopic appearance of PCGC or SRC.
Gastric carcinoma is classified as either early or advanced stage to help determine the appropriate type of intervention. Early gastric carcinoma is defined as invasive carcinoma confined to the mucosa and/or submucosa, regardless of lymph node metastases or tumor size. These tumors are generally smaller, measuring less than 5 cm in size, and found most commonly on the lesser curvature of the stomach at the angularis. Histologically, early gastric carcinoma will commonly present as well differentiated, mostly with tubular and papillary architecture. If the biopsies are composed of only mucosa, then distinguishing between well-differentiated carcinoma and carcinoma in situ or high grade dysplasia can be difficult. The presence of stromal desmoplasia in invasive carcinoma can help differentiate it from intramucosal invasion, which can contain single tumor cells within the lamina propria. This is an important distinction to make as intramucosal carcinoma does metastasize. Advanced gastric carcinomas will present grossly as either exophytic, ulcerated, or infiltrative tumors. Histologically, advanced gastric carcinomas will invade the muscularis propria and demonstrate cytologic and architectural heterogeneity, with a combination of patterns.
The 2010 World Health Organization classification determined four major histologic patterns of gastric cancer, which will often present with a combination of elements from the other patterns:
- Tubular: Most common pattern in early gastric carcinoma, with branching, distended or fused tubules containing intraluminal mucus, and nuclear and inflammatory debris
- Papillary: Most common in the proximal stomach with epithelial projections containing an underlying fibrovascular core. Also, it is frequently associated with liver metastases and an increased risk of lymph node involvement.
- Mucinous: Extracellular mucin makes up at least 50% of the tumor volume
- Poorly cohesive (including SRC): Mixture of signet ring and non-signet ring cells. Signet ring cells will have mucin pushing the nucleus to the periphery of the cell.
Helicobacter pylori (H. pylori) is a gram negative infectious bacteria that has been linked to gastric cancer. H. pylori is present in about half of the world’s population and other than gastric cancer, it is also associated with chronic gastritis, peptic ulcer disease, and gastric lymphomas. The bacteria is typically acquired during infancy and will remain for life if left untreated, with reactive oxygen species being generated that are capable of causing DNA damage due to the chronic infection. In addition, H. pylori can induce hypermethylation, resulting in the inactivation of tumor suppressor genes. Although H. pylori infection is considered a strong risk factor for developing gastric cancer, more commonly in intestinal type than diffuse type gastric cancer, only a small portion of those infected with the bacteria actually develop the malignancy. It is believed that approximately 80% of distal gastric cancers are due to a H. pylori infection, whereas there is little association between H. pylori and cardia gastric cancers.
In PCGC, such as this case, it is generally diagnosed in younger patients without a gender bias. Although PCGC can be associated with an H. pylori infection, it is more commonly related to a mutation in the tumor suppressor gene epithelial cadherin, also known as E-cadherin and CDH1. PCGC presents as an infiltrative growth of poorly differentiated, discohesive malignant cells that appear to arise from the middle layer of the mucosa. These cells can infiltrate as individual cells or as small clusters, but usually do not form glands (Image 2). If the gastric wall becomes extensively infiltrated by malignancy, the wall can be thickened and rigid, a macroscopic presentation termed as linitis plastica, which can lead to pyloric obstruction. Within PCGC, numerous signet ring cells can be present, leading to SRC. There is also a hereditary form of poorly cohesive gastric cancer referred to as hereditary diffuse gastric carcinoma, with an autosomal dominant pattern of inheritance. Histologically, it will include hyperchromatic nuclei, occasional mitoses, patchy intramucosal signet ring cells in the lamina propria, and carcinoma in situ associated with pagetoid spread of tumor cells along the preserved basement membrane. Hereditary diffuse gastric carcinoma will present with multifocal tumors under an intact mucosal surface, making diagnosis difficult. In patients with a CDH1 mutation and a family history of gastric carcinoma, a prophylactic gastrectomy is often the recommended treatment option.

References
- Adachi Y, Yasuda K, Inomata M, et al. Pathology and prognosis of gastric carcinoma well versus poorly differentiated type. Cancer. 2000;89(7)1218-24.
- Cancer. World Health Organization. Who.int. https://www.who.int/news-room/fact-sheets/detail/cancer. Published September 20, 2018. Accessed September 18, 2019.
- Carcas LP. Gastric cancer review. J Carcinog. 2014;13:14. Published 2014 Dec 19. doi:10.4103/1477-3163.146506
- Hu B, El Hajj N, Sittler S, Lammert N, Barnes R, Meloni-Ehrig A. Gastric cancer: Classification, histology and application of molecular pathology. J Gastrointest Oncol. 2012;3(3):251–261. doi:10.3978/j.issn.2078-6891.2012.021
- Mariette C, Carneiro F, Grabsch HI, et al. Consensus on the pathological definition and classification of poorly cohesive gastric carcinoma. Gastric Cancer. 2019;22(1):1-9 https://doi.org/10.1007/s10120-018-0868-0-
- Pernot S, Voron T, Perkins G, Lagorce-Pages C, Berger A, Taieb J. Signet-ring cell carcinoma of the stomach: Impact on prognosis and specific therapeutic challenge. World J Gastroenterol. 2015;21(40):11428–11438. doi:10.3748/wjg.v21.i40.11428
- Van Cutsem E, Sagaert X, Topal B, et al. Gastric Cancer. Lancet. 2016;388(10060):2654-64. https://doi.org/10.1016/S0140-6736(16)30354-3
- Weisenberg E. Diffuse (poorly cohesive) type carcinoma. Pathology Outlines. http://www.pathologyoutlines.com/topic/stomachdiffuse.html. Revised August 22, 2019. Accessed September 18, 2019

-Cory Nash is a board certified Pathologists’ Assistant, specializing in surgical and gross pathology. He currently works as a Pathologists’ Assistant at the University of Chicago Medical Center. His job involves the macroscopic examination, dissection and tissue submission of surgical specimens, ranging from biopsies to multi-organ resections. Cory has a special interest in head and neck pathology, as well as bone and soft tissue pathology. Cory can be followed on twitter at @iplaywithorgans.