Case History
The patient is a 6 year old who developed abdominal pain 2 days prior to admission. The patient was in school when the abdominal pain began, resulting in the patient doubling over in pain. The pain resolved within 1 hour, however, because the initial presentation was an unremitting abdominal pain, the patient was taken to an outside hospital for evaluation. There was no vomiting, diarrhea, or constipation. On physical exam, the patient was very tender to palpation in the right lower quadrant and was unable to tolerate deep palpation. A computed tomography scan was subsequently ordered which showed a large mass in the pelvic peritoneum. The patient was admitted to surgery for an exploratory laparotomy, with resection of the pelvic mass.
Diagnosis
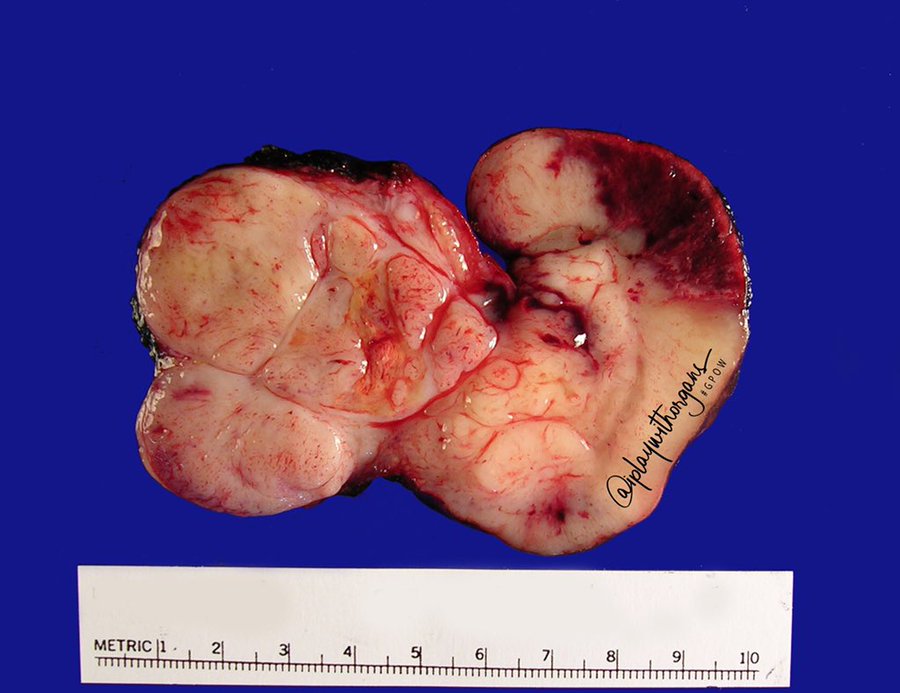
Received fresh in the Surgical Pathology laboratory is a 162.5 gm, 10.2 x 7.5 x 4.0 cm lobulated, ovoid mass of pink-tan, rubbery tissue that appears encapsulated by a thin translucent membrane. The margins are inked black and the specimen is serially sectioned revealing glistening, gray-tan soft tissue with focal areas of yellow discoloration and softening. Along one edge of the specimen, there is a 4.0 x 1.5 cm rim of dark red-brown, rubbery tissue (Figure 1). Portions of the fresh specimen are submitted in glutaraldehyde for electron microscopy if needed, RPMI for cytogenetics, and are snap-frozen as well. Touch preparations are also made and gross photographs are taken. Representative sections are submitted as follows:
Cassette 1-7: Sections of mass including inked capsule
Cassette 8-10: Representative sections from central portion of mass including areas of softening and discoloration
Cassette 11-13: Additional representative sections of the mass
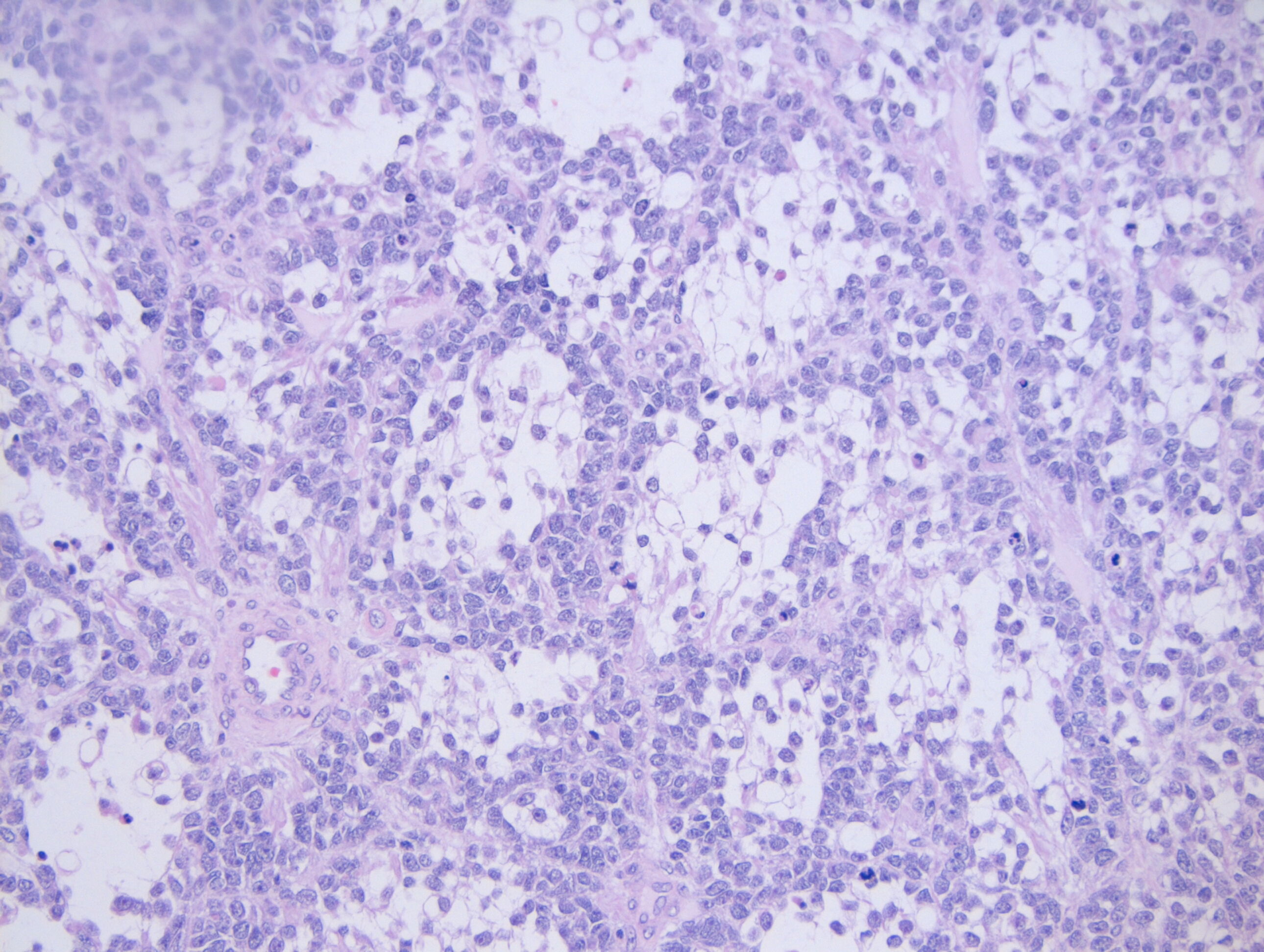
Histologically, the mass is composed of sheets and nests of small round cells along thin fibrous septa, giant multinucleated cells, and rare strap cells. Necrosis less than 5%. The margins are positive, although the specimen is unoriented. Venous and lymphatic invasion is absent. Immunohistochemical (IHC) stains are ordered and the results are listed below:
Positive IHC stains: Myogenin, desmin, CD56 and Bcl-2
Negative IHC stains: S-100, keratin AE1/AE3, CAM 5.2, SMA, CD99, Fli-1, WT-1, and EMA
In addition to the IHC stains, a portion of tissue was sent for cytogenetics testing, which showed a chromosomal translocation at t(2;13)(q35;q14). Based on the histologic appearance, IHC stains, and cytogenetic testing, the specimen was signed out as an alveolar rhabdomyosarcoma with a pathologic stageof pT2b, N0, MX.
Following the diagnosis, the patient was placed on a chemotherapy regimen of Vincristine, Adriamycin, Etoposide and Cytoxan, as well as radiation therapy.
Discussion
Rhabdomyosarcoma is the most common malignant soft tissue tumor in children and is the most common malignant solid tumor in children after neuroblastoma and Wilms tumor, accounting for 5-10% of all childhood tumors. 90% of these tumors occur in patients under the age of 25, and approximately 70% occur in children under 10 years of age. The most common locations of rhabdomyosarcoma are in the head and neck region, followed by the genitourinary system, extremities and then torso.
The 2013 World Health Organization classification of skeletal muscle tumors divided rhabdomyosarcoma into four types based on histology:
- Embryonal rhabdomyosarcoma (botryoides and anaplastic variant)
- Alveolar rhabdomyosarcoma (solid and anaplastic variant)
- Pleomorphic rhabdomyosarcoma
- Spindle cell/sclerosing rhabdomyosarcoma
Alveolar rhabdomyosarcoma (ARMS) accounts for approximately 20-30% of all rhabdomyosarcoma tumors, with no genetic predisposition. Although it is most common in teenagers, ARMS affects all ages. Most patients will present with a painless soft tissue mass, but based on the size and location of the mass, it may cause mass effect. A quarter of patients will have metastasis at the time of diagnosis, most commonly to the bone marrow, bones, and lymph nodes.
Grossly, ARMS presents as a solid, well-defined mass with a fleshy, tan-gray cut surface. Histologically, it is composed of small, blue, round cells and occasional round to spindle shaped rhabdomyoblasts. When compared to embryonal rhabdomyosarcoma, the rhabdomyoblasts in ARMS are slightly larger. ARMS is broken down into two subtypes: the classic subtype and the solid subtype. In the classic subtype, the tumor is composed of nests of cells that adhere to the edges of fibrous septa, resembling pulmonary alveoli (hence the name alveolar rhabdomyosarcoma). Multinucleated giant cells with a peripherally located nuclei may also be present. In the solid subtype, there will be nests and sheets of neoplastic cells that are separated by thin fibrovascular septa, but will not form in the classic alveolar pattern (Image 2).
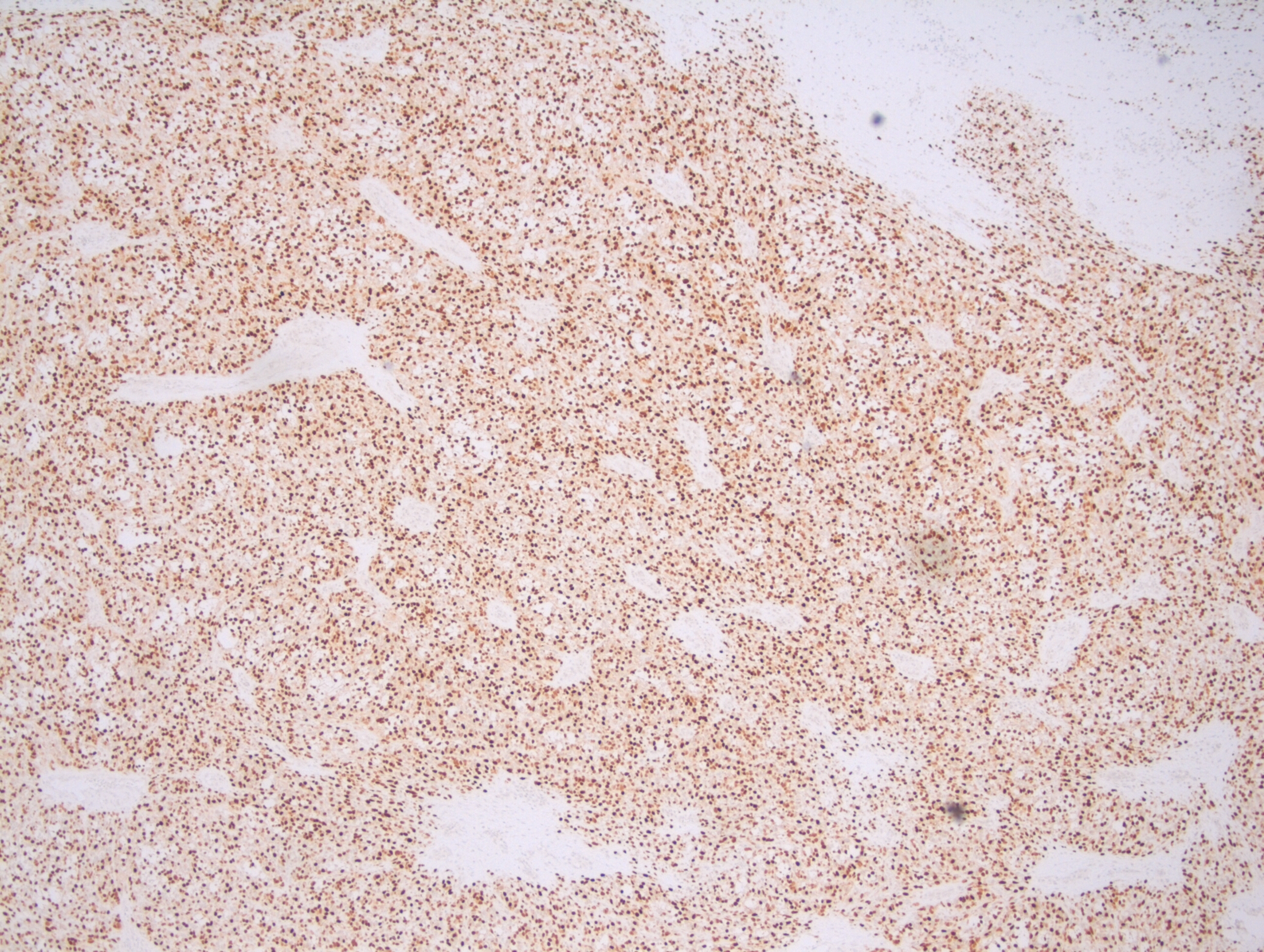
Due to the various appearances of rhabdomyosarcoma, it has become important to integrate immunohistochemical (IHC) stains and molecular testing into the diagnosis. The most common IHC stains that are used to determine the rhabdomyoblastic differentiation of a sarcoma is through the use of Myogenin and Myogenic differentiation 1 (MyoD1) stains, in which both stains will be positive in rhabdomyosarcoma. These two stains can be furthered used to help narrow down a diagnosis of ARMS because if more than 50% of the neoplastic cells express Myogenin, this is highly suggestive of a diagnosis of ARMS (Figure 3). In ARMS, the MyoD1 will have a variable expression. Additional positive IHC stains for ARMS can include: desmin, P-cadherin, and bcl-2.
To go along with IHC stains, molecular testing has been shown to be affective with determining the type of rhabdomyosarcoma. There have been two translocations that have been identified in ARMS. The first is at t(2;13)(q35;q14), which results in a fusion of the PAX3 gene with the FOXO1 gene (previously known as the FKHR gene). This translocation is present in 60% of all ARMS cases, and has been found to occur mostly in older children and younger adults. The second translocation is at t(1;13)(p36;q14), which results in a fusion of the PAX7 gene with FOXO1, and is present in approximately 20% of all ARMS cases. The remaining 20% are fusion negative, and are associated with the solid subtype histologically. There is early preliminary data that shows a less aggressive disease course in patients with the PAX7-FOXO1 fusion, compared to those with the PAX3-FOXO1 fusion.
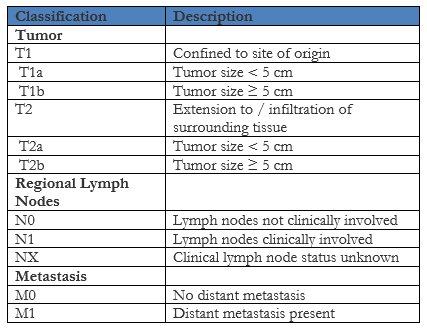
In order to determine the best treatment course, patients who are diagnosed with rhabdomyosarcoma are divided into a low risk, intermediate risk or high risk group based on the pathologic stage, clinical stage and clinical group. The pathologic stage is determined using the Pretreatment TNM Staging System that was set forth by the Intergroup Rhabdomyosarcoma Study (IRS) group (not the same as the TNM staging system put out by the American Joint Committee on Cancer) below:
The clinical stage is then determined using the TNM staging above and the Pretreatment Clinical Staging System below that is also put out by the IRS group:

In the above Clinical Staging System, a favorable site is defined as occurring in the orbit, biliary tract, head and neck region (excluding parameningeal) and genitourinary region (excluding prostate and bladder). Any other site not listed is considered unfavorable. Next, a clinical group is assigned based on the extent of the disease using the Clinical Grouping System below, which again is put out by the IRS group:
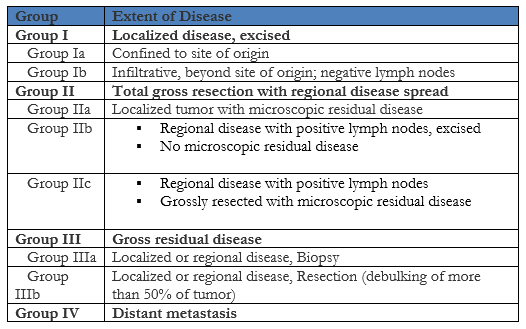
Lastly, based on the clinical stage and clinical group determined above, the patient is assigned a risk group of either low risk, intermediate risk, or high risk using the Children’s Oncology Group guidelines listed below:

When compared to embryonal rhabdomyosarcoma, which is the most common type of rhabdomyosarcoma, ARMS has a worst prognosis. The IRS group clinical group and stage can help to predict the overall outcome of the patient, with the standard treatment regimen composed of surgery, radiation therapy and chemotherapy.
References
- Dziuba I, Kurzawa P, Dopierala M, Larque A, Januszkiewicz-Lewandowska D. Rhabdomyosarcoma in Children – Current Pathologic and Molecular Classification. Pol J Pathol. 2018;69(1):20-32. doi:10.5114/pjp.2018.75333
- Liu H, Zhao W, Huang M, Zhou X, Gong Y, Lu Y. Alveolar rhabdomyosarcoma of nasopharynx and paranasal sinuses with metastasis to breast in a middle-aged woman: a case report and literature review. Int J Clin Exp Pathol. 2015;8(11):15316–15321. Published 2015 Nov 1.
- Owosho AA B Ch D, Huang SC Md, Chen S Mbbs, et al. A clinicopathologic study of head and neck rhabdomyosarcomas showing FOXO1 fusion-positive alveolar and MYOD1-mutant sclerosing are associated with unfavorable outcome. Oral Oncol. 2016;61:89–97. doi:10.1016/j.oraloncology.2016.08.017
- Ozer E. Alveolar Rhabdomyosarcoma. Pathology Outlines. http://www.pathologyoutlines.com/topic/softtissuealvrhabdo.html. Revised March 26, 2019. Accessed July 26, 2019.
- Rudzinski ER, Anderson JR, Hawkins DS, Skapek SX, Parham DM, Teot LA. The World Health Organization Classification of Skeletal Muscle Tumors in Pediatric Rhabdomyosarcoma: A Report From the Children’s Oncology Group. Arch Pathol Lab Med. 2015;139(10):1281–1287. doi:10.5858/arpa.2014-0475-OA
- Rhabdomyosarcoma Staging and Clinical Risk Groups. Stanford Medicine Surgical Pathology Criteria. http://surgpathcriteria.stanford.edu/srbc/rhabdomyosarcoma/staging.html. Accessed August 10, 2019

-Cory Nash is a board certified Pathologists’ Assistant, specializing in surgical and gross pathology. He currently works as a Pathologists’ Assistant at the University of Chicago Medical Center. His job involves the macroscopic examination, dissection and tissue submission of surgical specimens, ranging from biopsies to multi-organ resections. Cory has a special interest in head and neck pathology, as well as bone and soft tissue pathology. Cory can be followed on twitter at @iplaywithorgans.
