Case History
A 42 year old female with a history of neurofibromatosis, hypertension and Hashimoto’s thyroiditis had noted a mass on her forearm approximately 15 years ago. According to the patient, the mass did not change in size and did not cause her any discomfort during that time. Approximately 6 months prior to presenting to her primary physician, the mass began to increase in size and caused discomfort and pain. Upon examination with the Orthopedic Surgery department, a 20 x 20 cm firm, smooth mass on her forearm with mild pain on palpation was noted (Image 1). On MRI, the mass appeared to partially surround the radius and ulna, and encased the median, radial and ulnar nerves. A needle core biopsy was subsequently performed on the mass revealing a high grade malignant peripheral nerve sheath tumor (MPNST). A CT scan of the chest showed no evidence of metastatic disease. During her clinical visit, the use of neoadjuvant chemotherapy and chemoradiotherapy were discussed, but based on the large size of the mass, tumor response would have to be significant in order to allow for limb conserving surgery. At the time that the patient was seen, MPNSTs were not known to be chemosensitive and the chances of significant tumor response was very low (clinical drug trials have since shown some improvements in this area). In light of the poor response to systemic therapy of these tumors and the potentially toxic side effects of chemotherapy, the decision was made to proceed with amputation of the arm through the humerus.
Diagnosis
Frozen sections were sent from all the major peripheral nerves, including the ulnar, radial and median nerves. There was no evidence of any tumor consistent with a high-grade MPNST, although there was evidence of neurofibromas. There were atypical cells with hyperchromasia in the ulnar nerve margin, however, this was not considered to be consistent with a high grade MPNST. Received in the surgical pathology lab was an above elbow amputation consisting of a 30.0 cm long distal arm, an attached hand measuring 17.0 cm in maximum length., and a 4.5 cm long exposed humerus. The specimen is covered by grossly unremarkable skin, with a palpable mass in the mid-portion of the forearm. Sectioning reveals an 18.0 x 12.0 x 11.0 cm well-circumscribed mass composed of bulging, myxoid, white-tan tissue with central areas of hemorrhagic degeneration and yellow-tan friable tissue (Image 2). The bulging white-tan tissue is mainly found peripherally and encompasses approximately two-thirds of the mass. The mass is confined to a thin translucent lining and does not grossly invade neighboring soft tissue or overlying skin. The radial, median and ulnar nerves are adjacent to but not invaded by the mass, although the distal aspect of the mass shares a translucent, myxoid-like tissue with the peripheral nerve sheath of the ulnar and median nerves.
In addition to the standard bone and soft tissue margins that are taken, representative sections of the mass with the closest approach to the overlying skin are submitted. Sections demonstrating the relationship of the distal mass to the radial, median and ulnar nerves are submitted in separate cassettes. Lastly, representative sections sampled from various areas of the mass are submitted in an additional 15 blocks.
Histologically, the tumor consisted of spindle cells arranged in a fascicular pattern with intermittent whorled areas. The cells contained pleomorphic, hyperchromatic nuclei and intervening myxoid hypocellular areas. Mitotic figures were observed with sparse areas of necrosis and hemorrhage. S-100 was ordered on the prior biopsy of the mass, which was weakly positive. Based on these findings, the specimen was signed out as a malignant peripheral nerve sheath tumor.


Discussion
Malignant peripheral nerve sheath tumors (MPNST) are locally invasive tumors that are associated with medium to large nerves (as opposed to cranial or distal small verves) and commonly recur with eventual metastatic spread. Common sites for metastatic spread include lung, liver, brain, bones and adrenals. They are usually found in adults between the second and fifth decades of life, and account for only 5% of malignant soft tissue tumors. Approximately half of MPNSTs will occur sporadically, with the other half generally arising in the setting of neurofibromatosis type 1 (such as in this case). There is a high clinical suspicion for MPNST if the patient has a history of neurofibromatosis type 1 or if the tumor arises within a major nerve component.
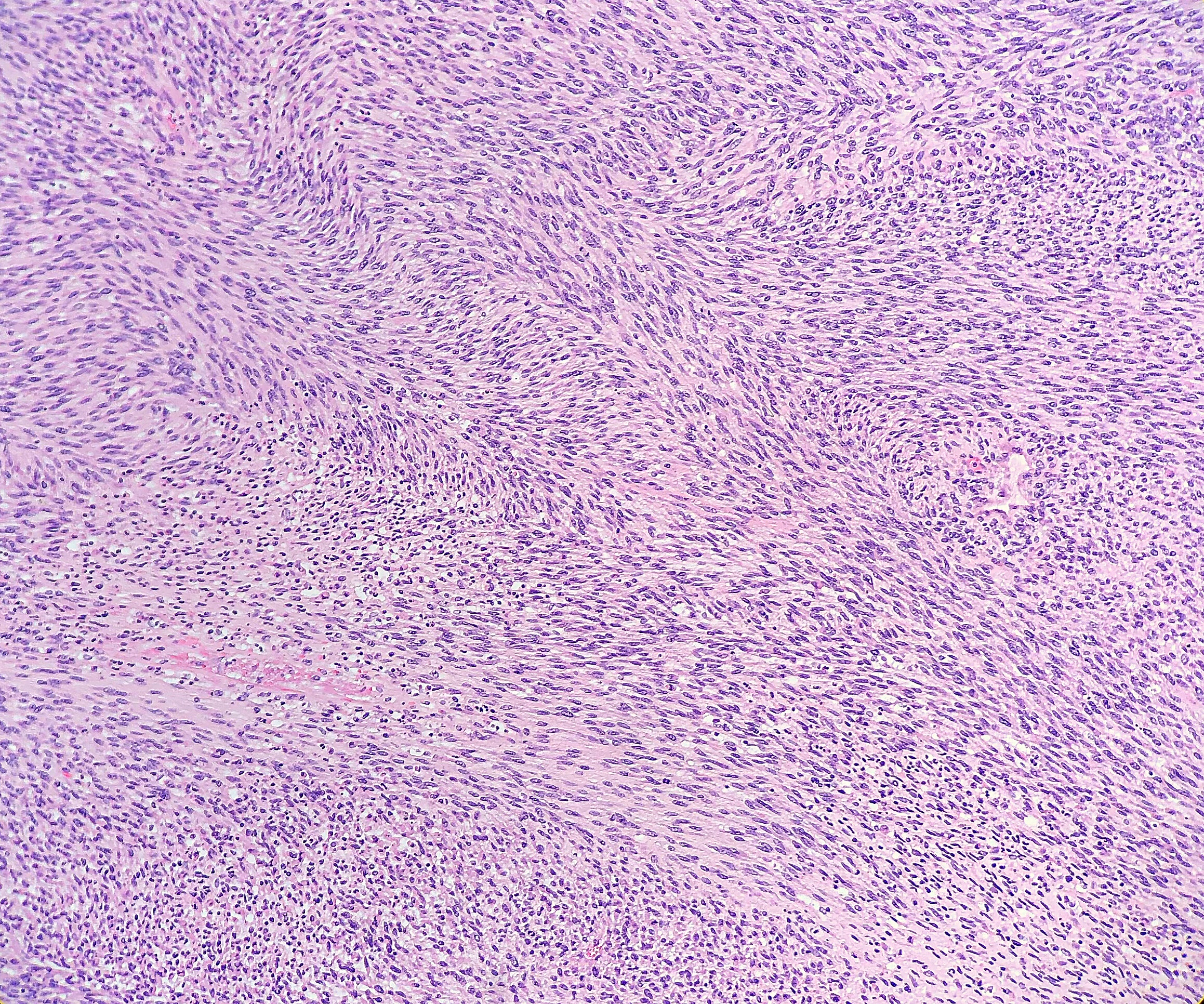
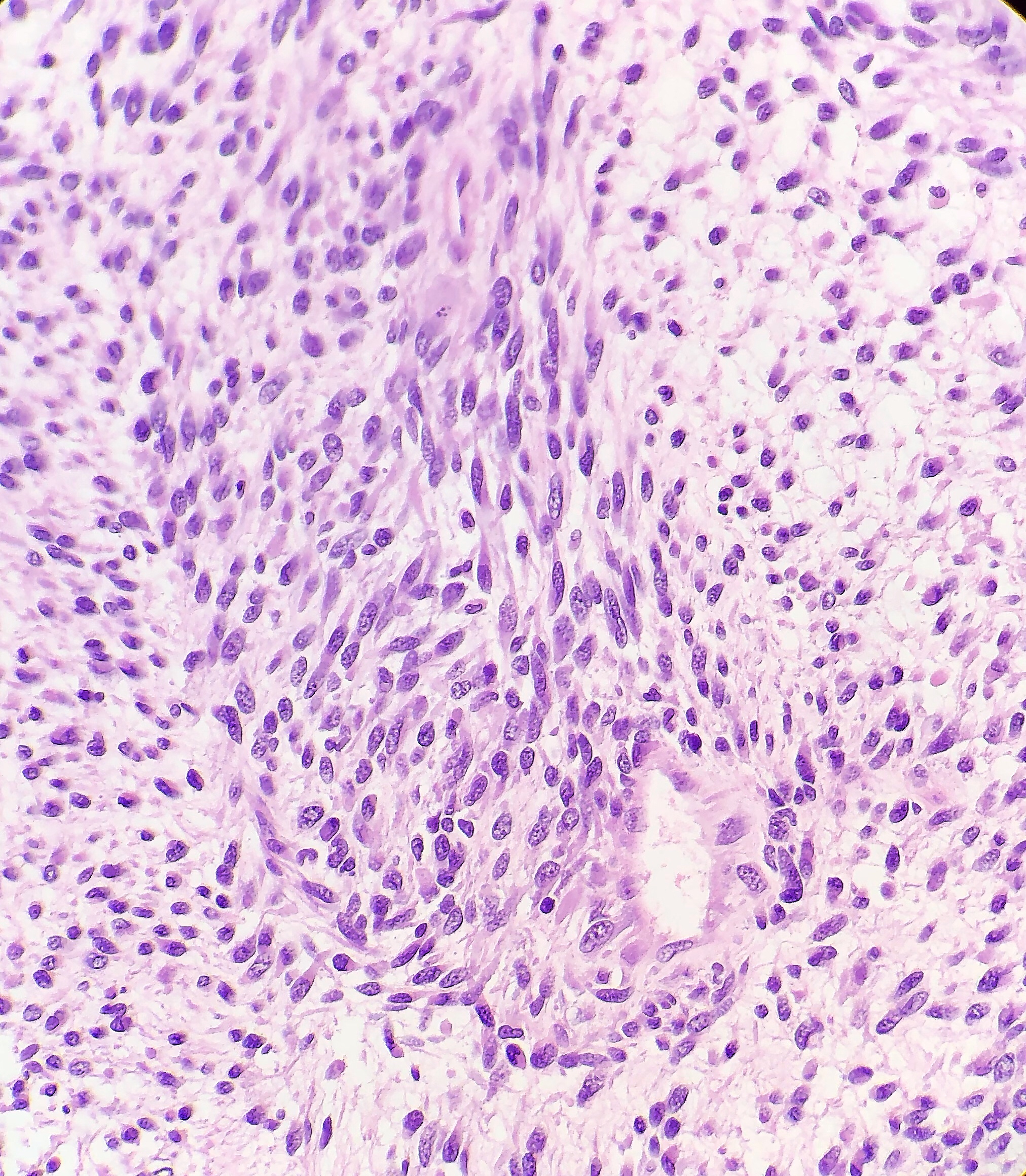
Grossly, MPNST will present as a large, poorly defined, fleshy tumor that runs along a nerve and involves adjacent soft tissue. Often, these tumors will have areas of hemorrhage or necrosis and can track along the length of a nerve. Histologically, the tumors are composed of monomorphic spindle cells arranged in fascicles, palisades and whorls, with compact comma-shaped, wavy or buckled hyperchromatic nuclei with alternating hypocellular foci. (Image 3 and 4). Mitotic figures and necrosis are common, and although S-100 is considered the best marker for MPNST, there is a lack of specificity and sensitivity for immunohistochemical markers. Due to the lack of immunohistochemical markers and molecular findings, as well as the variability associated with the cells, it has traditionally been difficult to diagnose MPNST. The differential diagnosis includes fibrosarcoma, monophasic synovial sarcoma, desmoplastic melanoma, and pleomorphic liposarcoma. Goldblum et al put forth the idea that a diagnosis of MPNST can be made if the tumor falls into any one of the following three categories:
- The tumor arises along a peripheral nerve
- The tumor arises from a pre-existing benign nerve sheath tumor, such as a neurofibroma
- The histologic features are consistent with a malignant Schwann cell tumor
Unfortunately, due to the aggressiveness of the tumor and high recurrence rate, MPNST has a poor prognosis with a 2 year overall survival rate of around 57% and a 5 year survival rate around 39%.
References
- Case of the week #443. Pathology Outlines. http://www.pathologyoutlines.com/caseofweek/case443.htm. Published November 15, 2017. Accessed March 10, 2019.
- Frosch MP, Anthony DC, De Girolami U. Malignant Peripheral Nerve Sheath Tumor. In: Kumar V, Abbas AK, Fausto N, Aster JC. Robbins and Cotran Pathologic Basis of Disease, 8th edition. Philadelphia, PA: Elsevier, Inc. 2010: 1341-1342
- Guo A, Liu A, Wei L, Song X. Malignant Peripheral Nerve Sheath Tumors: Differentiation Patterns and Immunohistochemical Features – A Mini-Review and Our New Findings. J Cancer. 2012; 3:303-309. http://www.jcancer.org/v03p0303.html. Accessed March 9, 2019.
- Hirbe AC, Cosper PF, Dahiya S, Van Tine BA. Neoadjuvant Ifosfamide and Epirubicin in the Treatment of Malignant Peripheral Nerve Sheath Tumors. Sarcoma. https://www.hindawi.com/journals/sarcoma/2017/3761292/cta/. Accessed March 10, 2019.
- Ramnani, DM. Malignant Peripheral Nerve Sheath Tumor. WebPathology. https://www.webpathology.com/case.asp?case=499. Accessed March 9, 2019.
- Shankar V. Malignant peripheral nerve sheath tumor (MPNST). Pathology Outlines. http://www.pathologyoutlines.com/topic/softtissuempnst.html. Revised September 12, 2018. Accessed March 9, 2019.

-Cory Nash is a board certified Pathologists’ Assistant, specializing in surgical and gross pathology. He currently works as a Pathologists’ Assistant at the University of Chicago Medical Center. His job involves the macroscopic examination, dissection and tissue submission of surgical specimens, ranging from biopsies to multi-organ resections. Cory has a special interest in head and neck pathology, as well as bone and soft tissue pathology. Cory can be followed on twitter at @iplaywithorgans.
