A 90 year old male is transferred from his nursing care facility to the hospital for management of acute appendicitis. He had acute onset of right lower quadrant abdominal pain the morning prior to admission with fevers, rigors and drenching sweats. Imaging showed ruptured appendicitis with a fecalith surrounded by small pockets of fluid. His past medical history included dementia, heart disease, hyperlipidemia, hypertension, and glucose intolerance. He denied having any prosthetic joints or valves. Blood was obtained for microbiological analysis.
Laboratory
Identification
Blood culture bottles flagged positive. Gram stain of the
blood culture bottles showed medium to long gram negative bacilli (Image 1).
The blood culture media was plated on blood, chocolate, and MacConkey agar. Aerobically,
yellow colonies grew on the blood and chocolate agar. The yellow colonies
turned red when exposed to 10% KOH (Image 2). Definitive diagnosis of Chryseobacterium gleum was obtained by
MALDI-TOF.
Image 1. Gram stain from the blood culture bottle shows gram negative bacilli.
Image 2. Growth of the organism on chocolate agar with addition of 10% KOH solution (circled in black).
Discussion
Chryseobacterium
gleum is a gram negative bacillus. They form yellow colonies that
grow on blood and chocolate agar. They rarely grow on MacConkey agar and are
non-fermenters when they do grow. Species of Chryseobacterium will turn red with addition of 20% KOH due to a pigment
protein called flexirubin. Interestingly, our lab had only 10% KOH and the
colonies turned red with this as well. Other key biochemical and physiologic
characteristics of Chryseobacterium
include being indole and oxidase positive and they are non-motile.
Chryseobacterium species
are found in the environment and are usually not part of normal flora, therefore
infection requires exposure of the bug to a debilitated patient in order to
colonize the respiratory tract. However, infection of other body sites that may
or may not have preceded respiratory tract colonization have been reported. These
organisms can survive in chlorinated tap water. They are an emerging cause of
hospital associated infections. No virulence factors have been studied. Risk
factors for infection include immunosuppression, trauma, surgery, burns,
foreign body implants and infused fluids. Of note, the patient was thought to
obtain his Chryseobacterium bacteremia
from his ruptured appendicitis.
For therapy, there are no definitive guidelines due to lack
of understanding of resistance mechanisms. These antibiotics have been reported
to have potential activity: Ciprofloxacin, rifampin, clindamycin,
trimethoprim/sulfamethoxazole and vancomycin (reportedly for C. indologenes). Our patient was given
Piperacillin/tazobactam, Ceftriaxone and metronidazole for two days, Cefepime
for one day, Vancomycin for a day. Infectious disease recommended continuing
piperacillin/tazobactam and starting trimethoprim/sulfamethoxazole and
discontinuing vancomycin.
Antimicrobial susceptibility testing was performed and
showed resistance to meropenem, aztreonam, gentamicin, and tobramycin. The
organism was susceptible to piperacillin/tazobactam and
trimethoprim/sulfamethoxazole.
Murray
P. Medical Microbiology. Seventh Edition. Elsevier; 2013.
Jain V, Hussain NAFA, Siddiqui T, Sahu C, Ghar M, Prasad
KN. Simultaneous isolation of Chryseobacterium gleum from bloodstream
and respiratory tract: first case report from India. JMM Case Rep.
2017;4(10):e005122. Published 2017 Oct 16. doi:10.1099/jmmcr.0.005122
-Angela Theiss, MD is a 3rd year anatomic and clinical pathology resident at the University of Vermont Medical Center.
-Christi Wojewoda, MD, is the Director of Clinical Microbiology
at the University of Vermont Medical Center and an Associate Professor
at the University of Vermont.
A 71 year old man with a history of multiple myeloma
presented with urinary incontinence and confusion and was found to have
hyperkalemia with renal failure. Imaging showed extensive inguinal
lymphadenopathy with concern for new lymphoma.
Excisional Lymph Node Biopsy
H&E 40x
Diagnosis
Sections
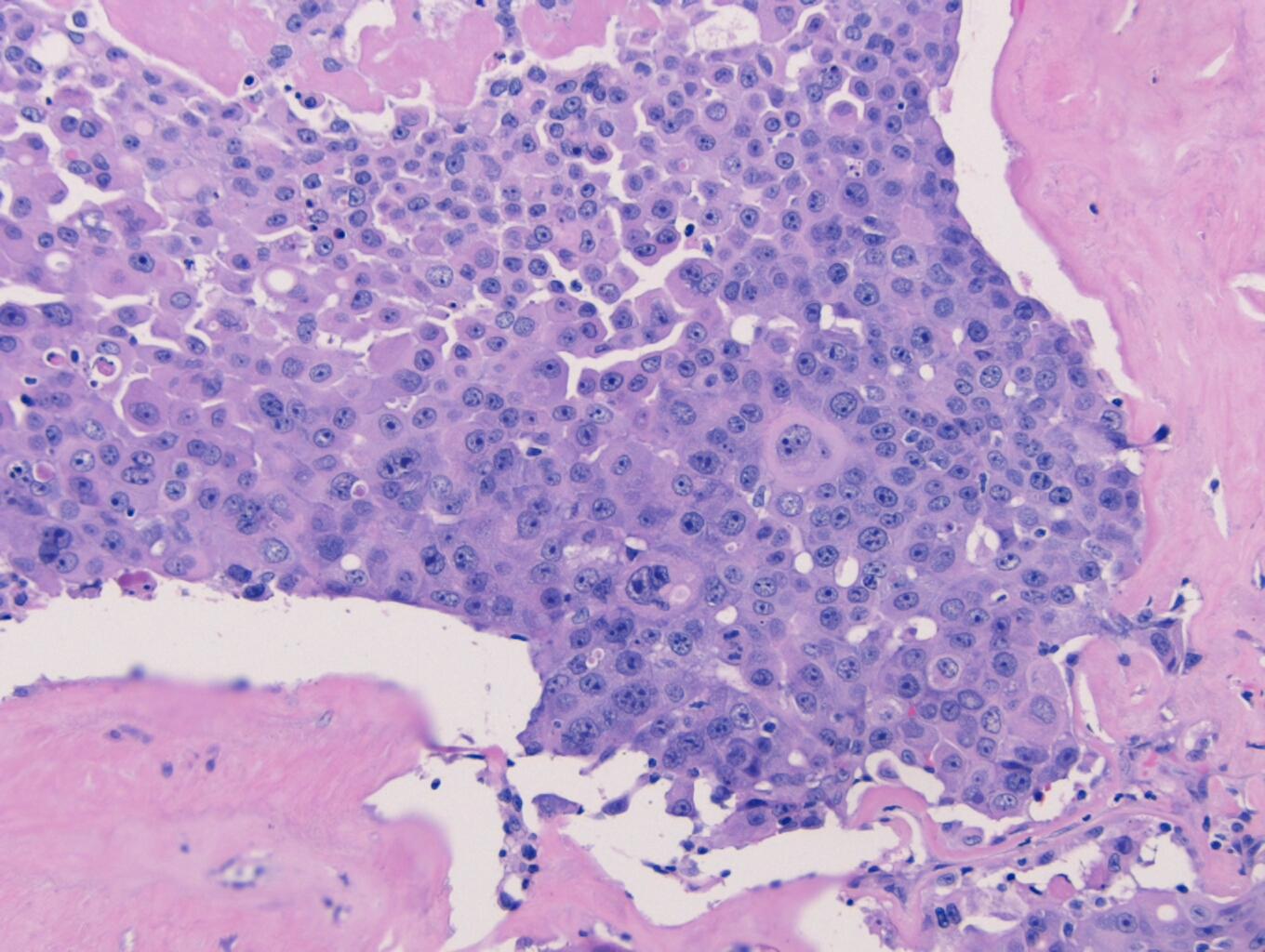
show an enlarged lymph node with complete effacement of the normal lymph node
architecture by sheets of medium and large plasmablastic cells. The cells have
round nuclear contours, large prominent nucleoli and moderate amounts of
amphophilic cytoplasm. Frequent apoptotic cells and scattered mitoses are seen.
Immunohistochemical stains show that the neoplastic cells
are immunoreactive for CD138, CD38, CD19 (dim) and MUM1. They are negative for
CD20, which highlights only small admixed B-cells. The cells are kappa
restricted by kappa and lambda immunostain. The Ki-67 proliferation index is
greater than 90%.
Taken together, the morphologic and immunophenotypic
features are of a high grade plasmablastic neoplasm. The differential diagnosis
includes plasmablastic myeloma and a plasmablastic lymphoma. Given the
patient’s history of a kappa restricted plasma cell dyscrasia, plasmablastic
myeloma is favored.
Discussion
Multiple myeloma is a neoplasm of clonal plasma cells that
accounts for 10% of all hematologic malignancies. It is most commonly seen in
adult and elderly patients with a male predominance. Plasma cells are generally
characterized by the presence of a “clockface” nuclei and distinct perinuclear
Hof or clearing of the cytoplasm containing a large number of Golgi bodies. The
morphology of plasma cell tumors can range from small mature plasma cells to
anaplastic or plasmablastic morphology. In this case, the cells showed
plasmablastic (PB) morphology, which is characterized by a large nucleus, large
nucleolus, fine reticular nuclear chromatin pattern, lack of nuclear Hof and
less abundant cytoplasm than typical plasma cells.1
The differential diagnosis for cases with this morphology primarily
includes PB lymphoma and PB myeloma with extramedullary involvement. PB
lymphoma is seen more commonly in HIV positive patients or patients with other
causes of immunodeficiency. It typically occurs in adults and has a male
predominance. The tumor generally presents outside of nodes and is most
frequently seen in the oral cavity/jaw. Patients tend to present with advanced
stage and bone marrow involvement. While PB lymphoma is categorized as a
distinct subtype of diffuse large B-cell lymphoma, PB myeloma is considered an
atypical morphologic variant of multiple myeloma and is treated with therapy
geared towards plasma cell neoplasms. 2
Making the distinction between these entities is difficult due to similarities in morphology and immunophenotype. Ultimately, the diagnosis is generally made based on the clinical context. In one series of “plasmablastic” neoplasms by Ahn, et. al., 6 out of 11 cases were called PB lymphoma, 2 out of 11 were called multiple myeloma and 3 were called indeterminate. Among the PB lymphoma patients, 4 were either HIV positive or had a history of immunosuppression. All 6 cases were positive for CD138 and negative for CD20 with EBV in situ hybridization positivity in 3 out of 6 cases. The multiple myeloma cases had evidence of end organ damage without lymphadenopathy. One indeterminate case had peritoneal nodules, lytic lesions and an EBV positive neoplasm in the bone marrow, which precluded a definitive diagnosis. 3
The immunophenotypic pattern seen in this case is typical of
these neoplasms and is characterized by the expression of plasma cell antigens (CD138,
CD38, MUM1) with either weak or negative expression of B-cell antigens (CD20). A
study by Vega et. al. looked at the immunophenotypic profiles in nine cases of
PB lymphoma and seven cases of PB myeloma. They found that the profiles were
nearly identical. All cases were
positive for MUM1/IRF4, CD138 and CD38 and negative for CD20, consistent with a
plasma cell immunophenotype. PAX5 and BCL6 were weakly positive in 2/9 and 1/5
PB lymphomas and were negative in all PB myelomas. A high Ki-67, overexpression
of P53 and loss of p16 and p27 were present in both tumors. There was no
evidence of HHV8 detected in either neoplasm. The presence of EBV-encoded RNA,
was seen in all PB lymphoma cases tested and negative in all plasma cell
myeloma cases. This was found to be statistically significant. 4
Unfortunately, both PB lymphoma and PB myeloma are aggressive
high grade neoplasms with a poor prognosis. A study conducted by Greipp et. al.
assessed the prognostic significance of plasmablastic morphology in a cohort of
patients from the Eastern Cooperative Oncology Group Myeloma Trial E9486. They
looked at bone marrow aspirates from 453 newly diagnosed multiple myeloma cases
in a 5 year period. Of the 453 aspirates, 8.2% were classified as PB
morphology. The overall survival of
patients with PB morphology was significantly shorter than patients with non-PB
morphology with a median of 1.9 years compared to 3.7 years. There did not
appear to be a relationship between PB morphology to other clinical or
laboratory features such as age, sex, bone lesions or type of M-protein. 5
References
M Srija, P Zachariah, V Unni, et. al.
Plasmablastic myeloma presenting as rapidly progressive renal failure in a
young adult, Indian Journal of Nephrology,
Volume 24(1): 2014, Page 41-44.
JJ Castillo, M Bibas, RN Miranda, The biology
and treatment of plasmablastic lymphoma, Blood,
Volume 125, 2015, Page 2323-2330.
J Ahn, R Okal, J Vos, et. al. Plasmablastic
Lymphoma vs Myeloma With Plasmablastic Morphology: An Ongoing Diagnostic
Dilemma, American Journal of Clinical Pathology,
Volume 144(2): 2015, Page A125.
F Vega, CC Chang, LJ Medeiros, et. al.
Plasmablastic lymphomas and plasmablastic plasma cell myelomas have nearly
identical immunophenotypic profiles. Modern
Pathology, Volume 18: 2005, Page 806-815.
PR Greipp, T Leong, J Bennett, et. al. Plasmablastic Morphology – An
Independent Prognostic Factor With Clinical and Laboratory Correlates: Eastern
Cooperative Oncology Group (ECOG) Myeloma Trial 39486 Report by the ECOG
Myeloma Laboratory Group, Blood, Volume 91: 1998, Page 2501-2507.
–Chelsea Marcus, MD is a Hematopathology Fellow at Beth Israel Deaconess Medical Center in Boston, MA. She has a particular interest in High-grade B-Cell lymphomas and the genetic alterations of these lymphomas.
This generation is very new to the workforce. In fact, the majority has not had a job yet as they are all eighteen and younger at the time of this writing. However, it is important to know how to adapt to this generation as they are starting to enter the workforce and many people communicate with this generation daily on a personal level.
This generation experiences a tremendous amount of uncertainty in their early lives. From the economic downturn in the late 2000s and school and concert shootings, this generation cares about security. This security is important on both a physical but also on a professional level; they want to make sure that they have professional stability. They care about making a difference, but not to the extent of Generation Y, the Millennial Generation.
There is some concern about this generation’s ability to connect with people on a long-term social level, mainly due to technological and social media advances. However, they do have a preference for face-to-face communication, so even if they do not come with that skill to the workplace, they can learn and adapt to it. Additionally, they are competitive and good multitaskers. They also have an entrepreneurial and independent spirit; they want to be in charge of their own projects and start their own companies. They are also looking into different ways to get their education that do not involve higher education and student debt. They are an imaginative generation with an intellectual curiosity.
Generation Z is the most diverse and open-minded generation, which means that they bring a plethora of ideas, background, concepts, and experiences. Leaders can utilize their diverse base to foster diversity of thought, practice, and skills at organizations. Including this generation as interns and entry-level workers is a good start to begin the process of mentoring this generation while learning from everything they bring to the organizational table.
-Lotte Mulder earned her Master’s of Education from the Harvard Graduate School of Education in 2013, where she focused on Leadership and Group Development. She’s currently working toward a PhD in Organizational Leadership. At ASCP, Lotte designs and facilitates the ASCP Leadership Institute, an online leadership certificate program. She has also built ASCP’s first patient ambassador program, called Patient Champions, which leverages patient stories as they relate to the value of the lab.
Ann Marie Nelson, MD has been a long-time hero of mine
from afar. If you don’t know who she is and what she has done, then after
reading this interview – you will see why! She is brilliant, selfless, kind
hearted, and is simply an inspiration!
Dr. Nelson is an anatomic and clinical pathologist with
more than 30 years’ experience in global infectious disease pathology and is
committed to improving health care by promoting timely and accurate diagnoses,
especially in parts of the world where resources are limited. She is currently
Infectious Disease Pathology Consultant at the Joint Pathology Center and
Professor of Pathology (visiting) at Duke University. The focus of her work has
been in HIV/AIDS pathology in the US and in sub-Saharan Africa. Currently she
works on educational projects and capacity building in anatomic pathology, and
linking anatomic pathology to ongoing clinical and epidemiologic research. She
is a founding member of InPaLa (International Pathology and Laboratory
Medicine), ASAP (African Strategies for Advancing Pathology) and serves as
co-chair of the subcommittee on education for the ASCP Partners in Pathology
initiative.
Recently, I had the good fortune of meeting her in person
and we sat down to talk about her amazing life and career, and what she
continues to do to contribute to the world.
Q:Your entire career has been focused on
improving the lives of others, through helping people get the care they need by
improving access, education, and opportunity. What inspired you to pursue a
career in global health in the first place especially as it relates to working
through pathology?
A: I’ve always
had a desire to travel even since I was young – I thought I wanted to do
something involving travelling – something like photography. When I was older,
I worked as a medical technician and the pathologist I worked under advised me
to pursue medical school. It was Vietnam war time though, so the odds of going
to medical school were 30:1 in California – but an opportunity arose to go to
medical school in Guadalajara, Mexico. I did, and this was my first time living
outside of the country. While there, I would participate in medical outreach
projects orchestrated by the medical school to serve the rural community
members. Naturally, since I was a Med Tech, I would run the laboratory
point-of-care diagnostics for the outreach. We would screen for parasites for
example, and this got me interested in infectious diseases.
I thought at first, I would pursue pediatrics, but pathology
drew me in. In 1979, I took a course in ‘Parasites for Medical Technicians’ and
met the folks in Tropical Medicine at UCLA. I met Dr. Marietta Voge, who had written a
book in Parasitology, and she became a mentor to me. Also, at the course, there
was a pathologist named Dr. Daniel Connor, from the AFIP [Armed Forces
Institute of Pathology], who was the editor of the ‘Atlas of Pathology of Tropical
and Extraordinary Diseases’. He gave a
lecture on his fascinating work which took place all around the world, but at
length in Uganda, and this was the inspiration for me. I thought “that’s what I want to do!”. Dr. Connor would become like a father
figure to me, and to this day my son calls him Grandpa. He has always been an
important supporter and mentor throughout my career.
Fast forward, I finished my residency training in pathology
and had the opportunity to spend four months at the AFIP working with the Infectious
disease pathology department. A few months later, they invited me to take a job
with them – which I did.
One of the hospitals in Africa that the AFIP supported was
the Karawa hospital in the Ubangi territory in the former Zaire. I worked for a
few months in the hospital there. While there, I met an African physician who
had just returned from completing his master’s in public health at Tulane
University. His name is Sambe Duale – I am now married to him. [She said this point with a smile and we both giggled
at how charming this story was!]
Towards the end of my work at Karawa, I was asked to help bring
pathology services to Kinshasa in a collaboration with the NIH, CDC, and the
Tropical Medicine Institute of Antwerp to work on Project SIDA [the first
project on AIDS in Africa]. I began working with Jim [James] Curran, Tom Quinn,
and Peter Piot, who were some of the people leading the project. I worked at the
Medical School in the Department of Pathology from the fall of 1986, and
continued to work there until 1991 when we were evacuated
out [by the US government due to the civil unrest that brought violence to
the capital].
After that, I continued to work in infectious disease pathology in the US, waiting for my son to graduate from high
school before considering working abroad again. In that time, I continued to be
heavily involved in IAP [International Academy of Pathology], working to
organize meetings, and contributing to building educational systems. I have given
world-wide lectures in at least 23 countries, in all continents except for
Antarctica. I retired from full time practice in 2015.
After my son graduated from high school, I decided to work
in Africa again on a Fulbright in Tanzania and Uganda. Professor Nelson
Sewankambo, who was the head of Makerere
University College of Health Sciences, invited me to mentor the
young pathologists at Makerere University. Robert Lukande was one of them – he
is now Chair of the pathology department there. We worked and wrote several
papers together, focusing on AIDS and autopsy. I gave lectures to multiple
departments, mentored staff, and made connections. I went and built
partnerships with everyone I could. You have to just go and talk to people, and
ask them “What can we do?”
Q:You worked to conduct a landmark survey of African pathologists to determine the status of pathology
resources in Sub-Saharan Africa. What were some of the key findings and how did
you collect all this data?
A: The idea for
the survey came when I was in Victoria Falls, South Africa for a pathology conference,
when I was speaking with Martin Hale. The realization that most of the conference
presenters were foreign pathologists, not African pathologists, struck us. We
who had been working in Africa knew the answer as to why – there weren’t enough
African pathologists. But there wasn’t any data, nobody knew how bad the
situation really was. The idea evolved over the next decade, Dan Milner helped
to put together an on-line survey that was translated into French and
Portuguese. When we finished the survey
in 2014, there were less than 800 pathologists in Sub-Saharan Africa.
The question then became, why aren’t
there more African pathologists? How
do you advocate for this to improve?
The data was largely based on person to person connections.
We had to reach out individually, involving people who spoke multiple
languages, made phone calls, sent emails…we worked for hundreds and hundreds of
hours. You have to really just get out on the street and talk to people.
This was the starting point so that we could measure
improvement. We are now working to update the survey and measure the progress
that’s been made.
Q:I’ve heard you have the nickname “Mama” in
and outside of Africa. How did this come about?
A: In 2006, it was the 100th anniversary of the IAP, and there was a pathologist from Nigeria who I had known, and he unofficially crowned me the “Mother of African Pathologists.” It stuck because people still refer to me as “Mama.” [Dr. Nelson told this story with a warm smile, and it was clear that this designation is an honor for her – I can easily tell that it is her kind soul and motherly nature that make people feel trust in her – “Mama” is absolutely a perfect fit.]
-Dana Razzano, MD is a Chief Resident in her third year in anatomic and clinical pathology at New York Medical College at Westchester Medical Center and will be starting her fellowship in Cytopathology at Yale University in 2020. She was a top 5 honoree in ASCP’s Forty Under 40 2018 and was named to The Pathologist’s Power List of 2018. Follow Dr. Razzano on twitter @Dr_DR_Cells.
The infectious disease service was consulted on an 81 year old female for persistent fevers. She initially presented a few weeks prior with cough & shortness of breath which was diagnosed as an acute chronic obstructive pulmonary disease (COPD) exacerbation for which she received levofloxacin and steroids. The patient continued to have a persistent cough and dysphagia after discharge. Her respiratory status and cough worsened and she was readmitted and intubated. Vancomycin, piperacillin/tazobactam and levofloxacin were started as well as fluconazole for suspected esophageal candidiasis. Her past medical history was significant for breast cancer, atrial fibrillation, and diabetes mellitus. Of note, patient was originally from Puerto Rico but moved to the United States 40 years ago and denied recent travel and any known tuberculosis exposures. She formerly worked in a deli packing cheeses. A bronchoscopy was performed and a brochoalveolar lavage (BAL) specimen as well as blood and stool specimens were submitted for bacterial culture and ova and parasite exam.
Laboratory
Identification
Image 1. Multiple larval forms in the stood specimen from an ova and parasite exam. (Iodine stain, 100X).
Image 2. High power of the larvae with a short buccal cavity (red arrow) and prominent genital primordium (blue arrow), (Iodine stain, 1000x).
The bronchoscopy revealed a bloody fluid admixed with clots
which was clinically consistent with diffuse alveolar hemorrhage. The
roundworms depicted above were identified in both the BAL and stool O&P
exam. Based on the presence of the short buccal cavity and the prominent
genital primordium and the absence of eggs, the identification of Strongyloides stercoralis was made.
Given the large amount of larvae present in both the lungs and gastrointestinal
tract, the patient was diagnosed with a strongyloidiasis hyperinfection.
Discussion
Strongyloides
stercoralis is classified as a nematode (roundworm) and is the cause of strongyloidiasis
in humans. The helminth is found worldwide, especially in warm climates and
underdeveloped countries, and is the cause of 30-100 million infections. Infection
is due to fecal contamination of soil, where free-living forms are found, or
water. Infective filariform larvae penetrate intact skin, particularly bare
feet, resulting in infection. The free living cycle begins with the
rhabditiform larvae passed through the stool develops into the infective
filariform larvae or when the
rhabditiform larvae mature into free living adult male & female
forms that mate and produce eggs which then hatch and become infective filariform
larvae that can infect humans. The parasitic life cycle begins with the
infective filariform larvae penetrates human skin. The worm is then either
coughed up from the lungs and swallowed or migrates to the small intestine
where eggs are laid and hatch.
Patients may present with gastrointestinal symptoms such as
abdominal pain, bloating, and diarrhea, pulmonary symptoms like dry cough and throat
irritation, or skin rashes along points of entry (feet, ankles). When the
larvae are in the lung, Loeffler’s syndrome, characterized by pneumonia
symptoms with coughing and wheezing, may develop due to an accumulation of
eosinophils in response to the parasitic infection. In patients who are
immunocompromised, the rhabditiform larvae can develop into the filariform
larvae in the host and can directly penetrate the bowel mucosa or perianal skin
resulting in autoinfection, dissemination throughout the body, and high
parasite burden. Symptoms of hyperinfection include bloody diarrhea, bowel
perforation, destruction of lung parenchyma with bloody sputum, meningitis, and
septicemia. Hyperinfection most commonly occurs after steroid administration
for asthma or COPD exacerbation, but can also be seen in those receiving
chemotherapy or who have had organ transplants.
In the laboratory, the diagnosis of S. stercoralis is most often made by an ova and parasite exam of
the stool, duodenal fluid, sputum or BAL specimens (Image 1). Most commonly the
rhabditiform larvae are present and are identified by the presence of a short
buccal cavity and prominent genital primordium (Image 2). These two features
are helpful in distinguishing S.
stercoralis from hookworms (Ancylostoma
spp. and Necator americanus) which
have a longer buccal cavity and indistinct genital primordium. The eggs of
these two nematodes are also very similar, although typically S. stercoralis eggs hatch before they
are passed in stool specimens. S.
stercoralis can also be visualized on H&E histology sections in the
crypts of intestinal biopsies where the adult female measures up to 2.2 mm in
length. Finally, serologic testing can be helpful when there is a high
suspicion of disease in the face of multiple negative stool exams, but cannot
distinguish between a current or past infection.
Most patients do not remember a specific
exposure and prevention includes wearing gloves and shoes when handling or
walking on soil that may contain contaminated fecal material. Treatment options
for an acute or chronic S. stercoralis include
a short course of ivermectin or albendazole. In the case of disseminated infection,
ivermectin should be given until stool and sputum exams are negative for 2
weeks. In the case of our patient, she was started on ivermectin, but succumbed
to the disease due to extensive pulmonary hemorrhage.
-Jaswinder Kaur, MD, is a fourth year Anatomic and Clinical Pathology resident at the University of Mississippi Medical Center.
-Lisa Stempak, MD, is an Assistant Professor of Pathology at the
University of Mississippi Medical Center in Jackson, MS. She is
certified by the American Board of Pathology in Anatomic and Clinical
Pathology as well as Medical Microbiology. She is the Director of
Clinical Pathology as well as the Microbiology and Serology
Laboratories. Her interests include infectious disease histology,
process and quality improvement, and resident education.
Welcome back. Last month I
talked about a colleague of mine, a fellow student who’s pursuing a career
in pathology. The month before that I wrote a bit about Just
Culture and how those of us in laboratory medicine ought to act as leaders
for patient advocacy—especially when it comes to putting the needs of patients
first. And in the spirit of progressing career timelines and fortuitous
transitions, this month I want to talk about a place where Just Culture is tangible,
where “patient come first” is a mission statement, and where I just spent the
last month rotating in their Department of Laboratory Medicine and Pathology:
The Mayo Clinic.
Image 1. Commemorative statue of the Mayo brothers in a park in front of the main building of the downtown campus at Mayo Clinic in Rochester, MN.
Before I go any further, if you haven’t seen the PBS Ken Burns’ documentary,
I highly suggest you do; it’s
fantastic. There are also a few excellent books on the hospital’s history and
vision here
and here.
But back to the rotation: I can’t express how lucky I feel having spent time
there or convey how much of a privilege it was to see pathology in a uniquely
Mayo way. What I can do is try to talk a little bit about my experience and
what that translates to regarding a culture of advocacy and collaboration; and
I’ll share a case conference I presented on my last day in a topic I find
fascinating.
Image 2. Ken Burns presents The Mayo Clinic: Faith, Hope, and Science on PBS, which aired September 2018.
Mission, Vision, and
Values
As with any hospital, academic center, clinic, etc., you’re
always going to have a driving philosophy that anchors the values of that
particular institution. Some of my experiences in larger academic centers tout
their strides at the forefront of medicine and translational research, others
advertise that they treat the whole person body and spirit. Community hospitals
sometimes lean into their integral part of, well, their communities as a center
for trust and health. Sometimes institutions have specific populations to cater
to or work intensely with industry and boast strong contributions to medical
science. At the Mayo Clinic you’d be hard pressed to miss the message (in
various forms) that “The Patients Come First”—in fact that line from many years
ago comes from Dr. William Mayo delivering a commencement speech at a Rush
Medical College graduation. (I was so happy to see so many Chicago-Mayo Clinic
connections!)
Image 3. Dr. W. Mayo articulated the concept of patients’ “needs come first” in a graduation speech at Rush Medical College in Chicago on June 15, 1910.
It becomes very obvious that this culture of advocacy
permeates into the daily proceedings there. The hospital makes a strong point
to celebrate outreach, education, and research; and clinicians are given a
cultivated environment in which to flex muscles of compassion for patient
outcomes. It makes you a better clinician, and I argue, person. Everyone at
this hospital has a voice and a seat at the table. I was continuously encouraged
to interact with staff, clinicians, residents, fellows, and patients and
contribute what I thought would benefit patient care. A unique perspective as a
visiting medical student with previous MLS experience was both noted and
celebrated.
Leadership in
Pathology
In many of my pieces on this blog, I frequently discuss how
we should champion active roles in testing stewardship, policy advocacy, and
promoting positive patient outcomes. Granted, when you find yourself in larger,
resource-rich, tertiary academic centers you can really push the envelope for
progress. But generally, those of us on the ‘scopes operate in this margin
between clinical medicine and translational research. Where does our leadership
come in? What does it look like? I think it comes in the form of prolific
contributions to societal guidelines and interdisciplinary work. Nowhere have I
seen this more than my month in Rochester.
So many of their residents contributed abstracts and
presentations at this year’s USCAP conference, some winning awards. The
academic cycle of producing something great requires strong support from your
home institution and that’s exactly what I saw. Not only were folks supported
for their trips to conferences per usual, they were celebrated—hallway handshakes,
accolades at morning conference, discussions post-meeting, and social media
shares. Which, by the way, social media is now a leadership staple. You can’t
go far in the present day without utilizing technology both inside and out of
your practice. The Pathologist
recently celebrated their first #TwitterPathAward for residents like Dr. Tiffany
Graham at UAB for contributions to medical education and advocacy in pathology.
Mayo clinicians, including residents, consultants, pathologist’s assistants,
and more share case studies, educational material, and cutting-edge pathology
news in terabytes! I now find myself increasingly active on social media
representing pathology and interests within our field.
Image 4. A spring 2015 issue of The Pathologist discussed the increasing presence of pathology in social media and the trends of utilization for medical laboratorians abound.
Side note: I’ve followed a number of these social media
pages about cases in pathology for a while, and when I was fortunate enough to
be part of ASCP’s Top 40 Under Forty 2017, I connected with lots of awesome
laboratorians. Some of which I got to meet this month! Including a fellow
blogger on this site, some celebrated path assistants, and a prolific
parasite-discussing clinical microbiologist.
Case Conference
So, my presentation was intense! I’ve given plenty of case
reports and conference discussions before, but this was an opportunity for me
to explore quite a rare case in genetics and connect it with my interests in
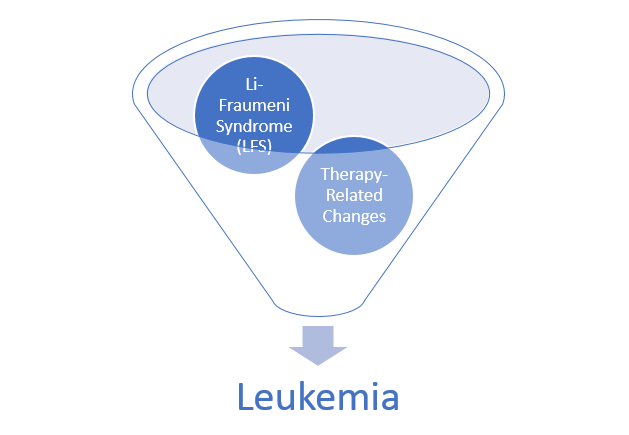
hematopathology. This was a case of a patient with Li-Fraumeni Syndrome (LFS)
who developed therapy-related Acute Myeloid Leukemia. It’s not a current case
and has since been signed-out and closed, but I’ll only be talking about the
pathologic entities involved.
Image 5. Remember that power of social media I mentioned earlier? Well, what better way to share information for other medical students interested in pathology and interested in visiting Mayo Clinic! Having my presentation grab an honorable mention amidst their productive and busy residents was great! #path2path #hemepath #lablogatory
Essentially, this patient was found to have Li-Fraumeni after
the second manifestation of an acute sarcoma—the first being osteosarcoma in
her teenage years and the second breast cancer in her 30s. Both cancer
diagnoses were treated accordingly, and this patient was going through routine
work-up for anemia before being referred to the Mayo Clinic. By the time the
patient reached there, the clinical investigation included a battery of testing
for causes of anemia—all within normal limits—so a bone marrow examination was
performed which revealed a significant, though not acute (<20% blasts),
myelodysplastic process. A follow-up in-house bone marrow collection revealed
hypercellular marrow, now in acute myeloid proliferation, with abnormal myeloid
cell maturation and very complex cytogenetics. She had a very complex karyotype
and several detectable mutations which were consistent with the WHO’s
classification and description of therapy-related myeloid neoplasm as a sequale
to the treatments she received for her prior cancers. In the setting of a
patient with LFS, it is almost impossible to avoid malignancy. The following
slides are a (very abridged) summary taken from my presentation of this
patient’s case:
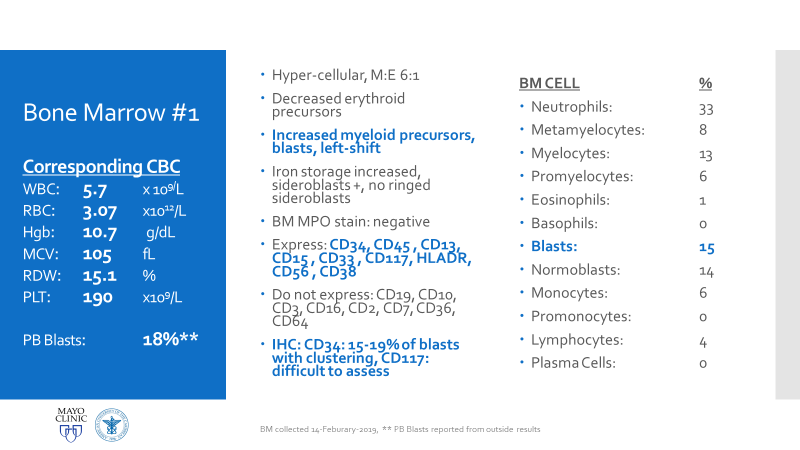
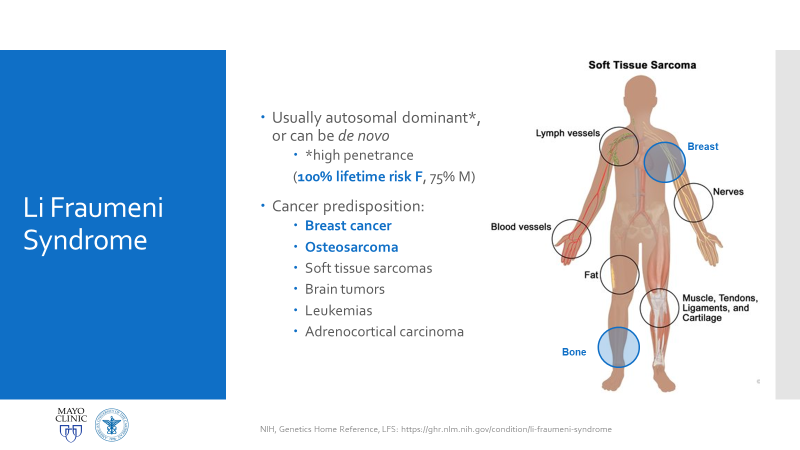
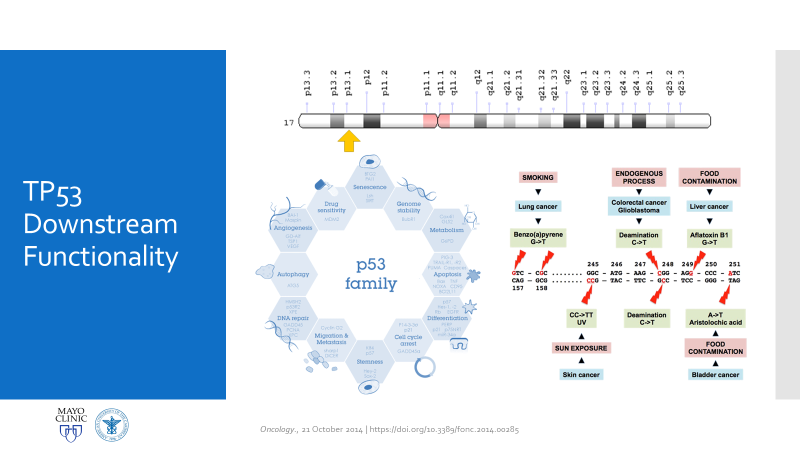
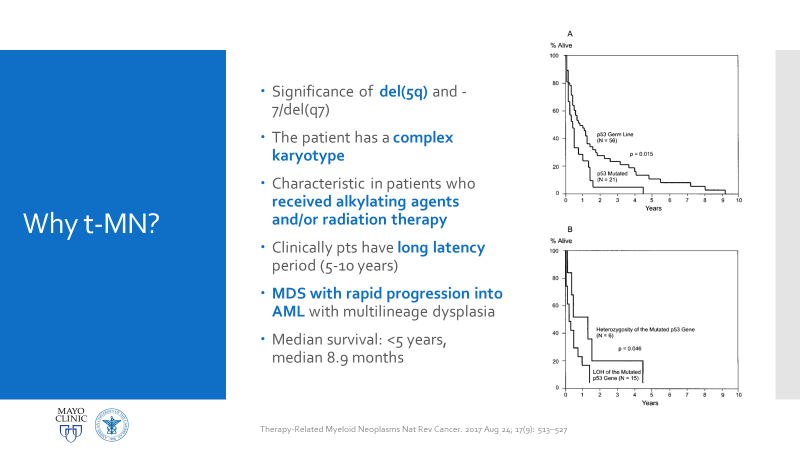
Figure 1. Official LFS and AML discussion. As mentioned, this is the case of a patient with a history of osteosarcoma and breast carcinoma, both treated, now presenting status-post initial work-up for evaluating possible causes for anemia. Ultimately, when reaching a bone marrow examination, certain myelodysplastic features were discovered, referring this case for close investigation and expanding the differential to include various hematologic malignancies.Figure 2. This bone marrow biopsy was evaluated at an outside institution and was reported to this patient’s case at Mayo Clinic. Note the presence of myeloid lineage blasts cells in the peripheral blood (PB) and bone marrow (BM) evaluations, however, at less than 20% this would not immediately indicate any acute myeloid crisis. There is a definitive left-shift in maturity with myeloid dysplasia. Figure 3. This bone marrow evaluation was done about a month after the previous reported one. Note the significant increase in myeloid blasts present in both peripheral and bone marrow specimens. This time, there was significant dysplasia noted in multiple lineages as well as particular changes in granulocytic lines including left-shift and pseudo Pelger-Huet cells present. This diagnosis was upgraded from myeloid dysplasia to acute myeloid leukemia in the setting of myelodysplasia. The blast count has now crossed the 20% threshold and there are marked changes to morphology in several cell lines. Hypercellularity and cytogenetic testing were also highly contributory in this diagnosis. Not included in this slide but CD34+ cells that previously expressed CD15, CD33, and CD38 were now negative for those three markers. This indicates decrease in maturity and a poorer prognostic and clinical assessment of this malignancy.Figure 4. A peripheral blood smear at the time of the second bone marrow specimen. In almost every field photographed, there were myeloid blast cells present. No Auer rods were seen, but many blasts had granules. There was left shift, and some immature granulocytes were present. Erythroid immaturity was demonstrated with morphology and circulating nucleated RBCs. Abnormalities in granulocytic lineages were present with hypogranular neutrophils and pseudo Pelger-Huet morphology.Figure 5. At nearly any age, this bone marrow needle core biopsy on H&E stain would qualify as hypercellular. At low to medium power this is clearly evident. At higher powers, note the presence of predominantly immature granulocytes (with very few, if any, mature PMNs) as well as numerous blasts—on H&E blasts appear differently, but appreciate the increased number of cells with active nuclei, condensing chromatin, and prominent nucleoli. Figure 6. Back to traditional hematology staining, you can still appreciate this bone marrow aspirate’s hypercellularity. There is a labeled megakaryocyte (which appears slightly abnormal) to scale against the numerous, immature and left-shifted granulocytes which overrun the fields. Myeloid blasts are seen in high numbers, with granules and prominent nucleoli. Increased levels of mitotic activity, abundant (and some abnormal) myeloid precursors, and a highly proliferative picture is appreciated.Figure 7. Li-Fraumeni Syndrome (LFS) is a rare genetic predisposition to soft-tissue sarcomas. It is a germline mutation of either TP53 or CHECK2, more often the former. The mutation usually has an autosomal dominant inheritance pattern and has very high penetrance, more so in females (possibly due to the fact that the most common presentation of tumor formation in LFS is breast cancer). Note that this patient had a clinical history significant for both breast carcinoma and osteosarcoma which were treated with chemotherapy and radiation.Figure 8a. Patients with LFS often have a germline mutation in p53, a very significant tumor suppressor gene, which is implicated in a wide host of cellular functions. Located on the short arm of Chromosome 17, when mutated this gene affects a myriad of pathways including cell senescence, growth cycle response, proliferation, DNA damage repair from mutations, epigenetic, or exogenous causes, and programmed cell death. If this downstream protection against severe DNA compromise is lost, this becomes a highly pre-cancerous environment for Knudsen’s “second hit” to negatively affect cells and ultimately lead to a vast array of malignancies. Figure 8b. I mean just look! P53 is a serious player in cell survival and DNA damage recovery. It is the archetype example of a tumor suppressor gene and is implicated in an ever-growing number of cell survival and growth cycle pathways—of course a loss of p53 function would set the stage for high-risk.Figure 9a. The World Health Organization (WHO) and its updated guidelines for diagnosing and addressing hematologic malignancies now includes a lot of new data regarding the molecular biology of cancer. Its applications to diagnostics in hematopathology are growing daily. In these guidelines, the WHO classify AML into seven general categories. For reasons relating to her clinical history of cancers and treatment, as well as the timeline she presented with, t-MN or therapy-related myeloid neoplasm would be an appropriate diagnosis. Figure 9b. The American Society of Hematology (ASH) and the College of American Pathologists (CAP) co-wrote guidelines for the diagnosis of AML and published a number of recommendations in The Hematologist in 2017-2018. Essentially, proper laboratory test utilization and incorporation with significant clinical history is crucial. Staying organized and operating within WHO guidelines for hematologic malignancy diagnosis is just as important. The ASH/CAP guidelines tell diagnosticians to think about several key questions when approaching AML which further underscores the values of consistency, efficiency, and appropriate utilization.Figure 10a. The reason for establishing a diagnosis of therapy-related AML is a significant one. The use of Topoisomerase II inhibitors, alkylating agents, antimetabolites, and radiation therapy all affect the genetic components relating to this particular leukemia. To correlate further, the patient had a 5q deletion, a complex karyotype, a history of receiving all treatments related to this entity, and a presentation of myelodysplasia which rapidly progressed to AML.Figure 10b. LFS can cause leukemia on its own, AML can present as a hematologic malignancy on its own too; but this patient’s clinical history and treatment history lean the diagnosis away from de novo cancer to a myeloid process in response to a latent treatment effect.
Why All of This
Matters
There are two main reasons why all of this is important
enough to discuss in a case conference. First, as clinicians from the bench to
the bedside we should all strive to talk through the toughest diagnoses and
share with each other what best practices, lessons, and goals we can reach
together. In the setting of Li-Fraumeni Syndrome it becomes critical to
evaluate new onset (especially myeloid) neoplasms. TP53 mutations are associated with the lowest survival rates in
acute myeloid leukemia, which has its own diagnostic and prognostic
classifications set forth by the World Health Organization. Furthermore,
understanding appropriate patient history, clinical information, and what
appropriate lab investigation means is crucial. It not only keeps the needs and
interests of the patient first, but also translates to the proper utilization
of resources for the best results in the best timelines. Potential future
implications of concurrent ongoing work in hematopathology and molecular
genetics may yield therapeutic and diagnostic benefits we are not yet aware
of—we must constantly include updates as we practice.
Second, this was an opportunity to share insights into the
diagnosis and discussion of AML that came from my clinical experiences before
rotating there. I previously mentioned the demonstrated value of including
clinical viewpoints for the benefit of patient care outcomes, so appropriately
I incorporated these topics into this case conference and included the
following points to consider:
Hematologic
premetastatic niches
When I was in graduate school at Rush
University in Chicago, I did some research in hematopoietic responses to
various therapies in the context of proliferation and understanding
mobilization for transplant and engraftment. In this work, I became familiar
with the concept of a reactive stroma and a “pre-metastatic niche.” There are
small microenvironments in which hematopoietic, mesenchymal, and endothelial
cell lines in the bone marrow thrive and develop which are full of cytokines
and cell-cell interactions. My work focused on mobilizing all three lines with
a CXCR4 target, but the concept holds true when considering germline and
somatic mutability. In effect, those cells with pre-malignant mutations can
cluster and affect the environment of other cells maturing in the same setting.
The same way invasive cells can break through barriers to metastasize and
spread past their in situ conditions,
the same mobilizing spread can grow from pre-metastatic clusters. This, again,
opens the discussion for treatment targets in future LFS and/or AML patients as
molecular pathology expands.
Acute
Myeloid Leukemia and Myeloid Sarcoma
In a recently published paper in Histopathology, I was part of a team at
the UAB hospital’s department of pathology which discussed their experience
with patients diagnosed with myeloid sarcomas (MS). The point was to look for
correlations with MS to connect the entity with age, sex, location of tumor,
AML status, genetics, etc. Ultimately, what became the highest predictor of
disease was a complex karyotype, consistent with other concurrent literature.
With respect to this patient, what if there was another soft tissue (or other
location) sarcoma alongside her myelodysplastic picture. What if she had a low
blast count, or hypocellular bone marrow, or necrosis/fibrosis, or had received
G-CSF? Would AML with myeloid sarcoma be considered in this diagnostic setting,
would myeloid sarcoma be something to worry about in her future or in her
clinical history as a misdiagnosis? The take-home message would be to pay close
attention to patient clinical history and stay both focused on the current
diagnostic work-up but also open enough to avoid pitfalls in diagnostic
challenges.
Misdiagnosis
in clinical settings
In a case report from 2017 I discussed a
patient who had bilateral lung nodules several years after being treated for
breast carcinoma. It was initially thought to be relapse but was later
correctly diagnosed as de novo
peripheral T-cell lymphoma (PTCL). This could have very well been the same
clinical scenario, with a different cell lineage. The lesson gleaned here is
the same as those ASH/CAP guidelines: stay organized, consistent, and
purposeful with your testing and investigation. What came down to a few
immunohistochemical markers in this PTCL case could make all the difference in
another case. Missing the clinical history and specific genetic mutations
present in this LFS/AML patient could have led to a diagnosis of a
myelodysplasia related AML instead of a therapy-related one, especially in the
setting of such a severe germline pre-disposition.
Future
plans for this patient
I thought it was ultimately important to
discuss the patient’s future plans with the audience. In pathology we often
sign-off after we sign-out. So, in order to make sure we emphasize the
patient’s best interests moving forward from a poor prognostic diagnosis, we
discussed her enrollment in a trial aimed at improving bone marrow donor
matching based on HLA and KIR combination typing. This a relatively new and
promising concept in the literature which I hold high hopes for.
If anything, this was something I learned last month: in
order for you to call the quality of care the highest possible, you have to
uphold many standards, both clinical and non-clinical. Clinically we all have
to share with each other the latest and greatest in modern literature and
advances in interdisciplinary or translational research. Aside from this,
however, we have to keep each other human and connected to our patients. I
never like to hear the stereotypes
in pathology that place us in lab medicine miles away from patient care;
instead, we do things every day that impact our patients’ lives greatly. And
when we keep ourselves connected to that fact, like the philosophy at the Mayo
Clinic, then we can boast our quality of care—from small community hospital to
academic trauma center. Because its not the size of the lens on the scope, it’s
the vast scope of impact we look through in a lens of compassion.
There you have it. That’s my month at Mayo and a case conference in a nutshell. It was a fantastic experience and I have to say it—I had a blast!
Thanks for reading, I’ll see you next time!
And have a Happy Lab Week 2019!
–Constantine E. Kanakis MSc, MLS (ASCP)CM graduated from Loyola University Chicago with a BS in Molecular Biology and Bioethics and then Rush University with an MS in Medical Laboratory Science. He is currently a medical student actively involved in public health and laboratory medicine, conducting clinicals at Bronx-Care Hospital Center in New York City.
A 63 year old man presented with a long standing history
of a recurring pleomorphic adenoma of the parotid gland. As a child, the
patient had radiotherapy to the bilateral parotid glands for parotid swelling. He
then developed a left parotid mass ~15 years later and underwent parotidectomy. After
another recurrence ~15 years after the initial parotidectomy, he underwent a
second resection of multiple masses in the preauricular region. The patient
then developed a recurrence ~20 years after the second resection and underwent
neutron beam therapy. The patient tolerated the treatment well noting mild dry
mouth, which is persistent, and left ear pain, but otherwise has no major
long-term sequelae from the treatment. Eighteen years after the neutron beam
therapy, the patient developed a left submandibular mass. A subsequent biopsy
of the mass revealed a pleomorphic adenoma. Enlarged left and right submental and
submandibular nodes were noted, with biopsies performed at an outside hospital
of these nodes demonstrating metastatic poorly differentiated carcinoma within
three lymph nodes. It was noted on this pathology report that the histological
features, in light of the history, could represent a carcinoma ex pleomorphic
adenoma. A CT scan of the head and neck revealed a large multiloculated, cystic,
rim-enhancing mass within the left parotid gland, as well as large enhancing
lymph nodes within the right anterior and posterior cervical triangle and the
right submandibular space, the largest of which measured 2.1 cm. A PET scan
showed increased activity within the right neck. Upon meeting with
otolaryngology, a 4.0 x 7.0 cm lobular, non-fixed left parotid mass, and two
level 1B right sided nodes, were palpated. Based on the patient’s history,
physical exam, and prior biopsy results, it was decided to proceed with a parotidectomy
and bilateral neck dissection.
Diagnosis
Received
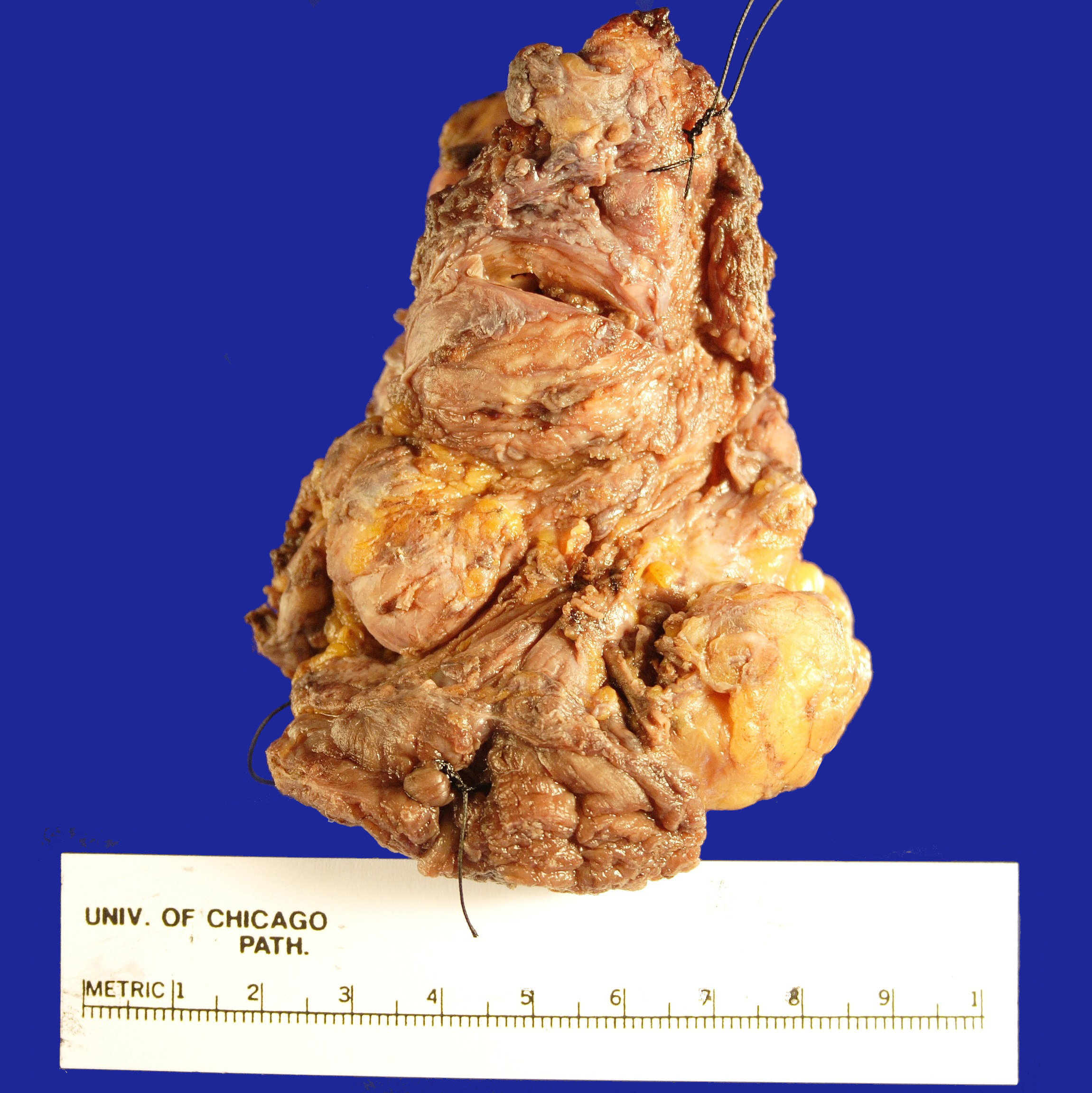
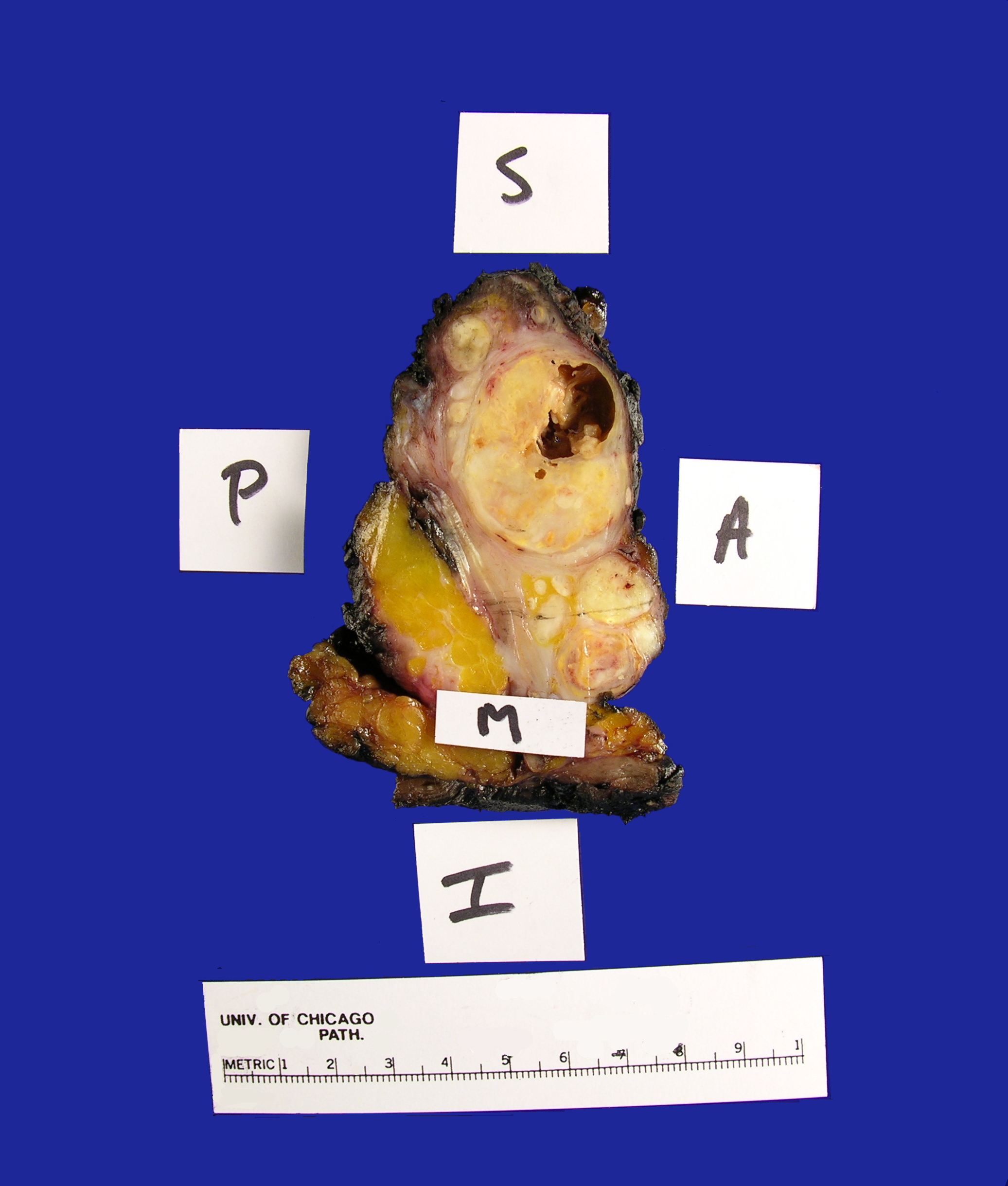
in the Surgical Pathology laboratory is a soft tissue mass resection from the
area of the left parotid gland measuring 9.0 x 6.0 x 4.2 cm. The specimen is
oriented by a single long stitch designating the superior aspect, and a double
long stitch designating the lateral aspect (Figure 1). The specimen is entirely
inked black, and then bisected to reveal multiple discrete, white-tan,
partially cystic masses ranging in size from 0.2-4.0 cm in greatest dimension and
measuring 7.0 x 3.5 x 3.0 cm in aggregate dimension (Figure 2). The largest
mass is partially cystic with the cystic component measuring 1.2 cm in greatest
dimension. This largest mass abuts the anterior, medial and lateral margins. The
remaining tumor deposits are located:
– 1.2 cm
from the inferior margin
– 0.4 cm
from the superior margin
– 0.9 cm
from the posterior margin
No gross
salivary gland tissue is identified. The remainder of the specimen consists of
unremarkable yellow adipose tissue and red-brown skeletal muscle. The specimen
is submitted as follows.
Cassette
1: superior margin
Cassette
2: representative sections of
anterior margin
Cassette
3: anterior superior margin
Cassette
4: anterior inferior margin
Cassette
5: posterior margin
Cassette
6-9: representative sections of mass
with approach to lateral margin
Cassette
10: representative sections of mass
with approach to medial margin
Cassette
11: mass in relation to
surrounding skeletal muscle
Cassette12-15: representative sections of mass
On
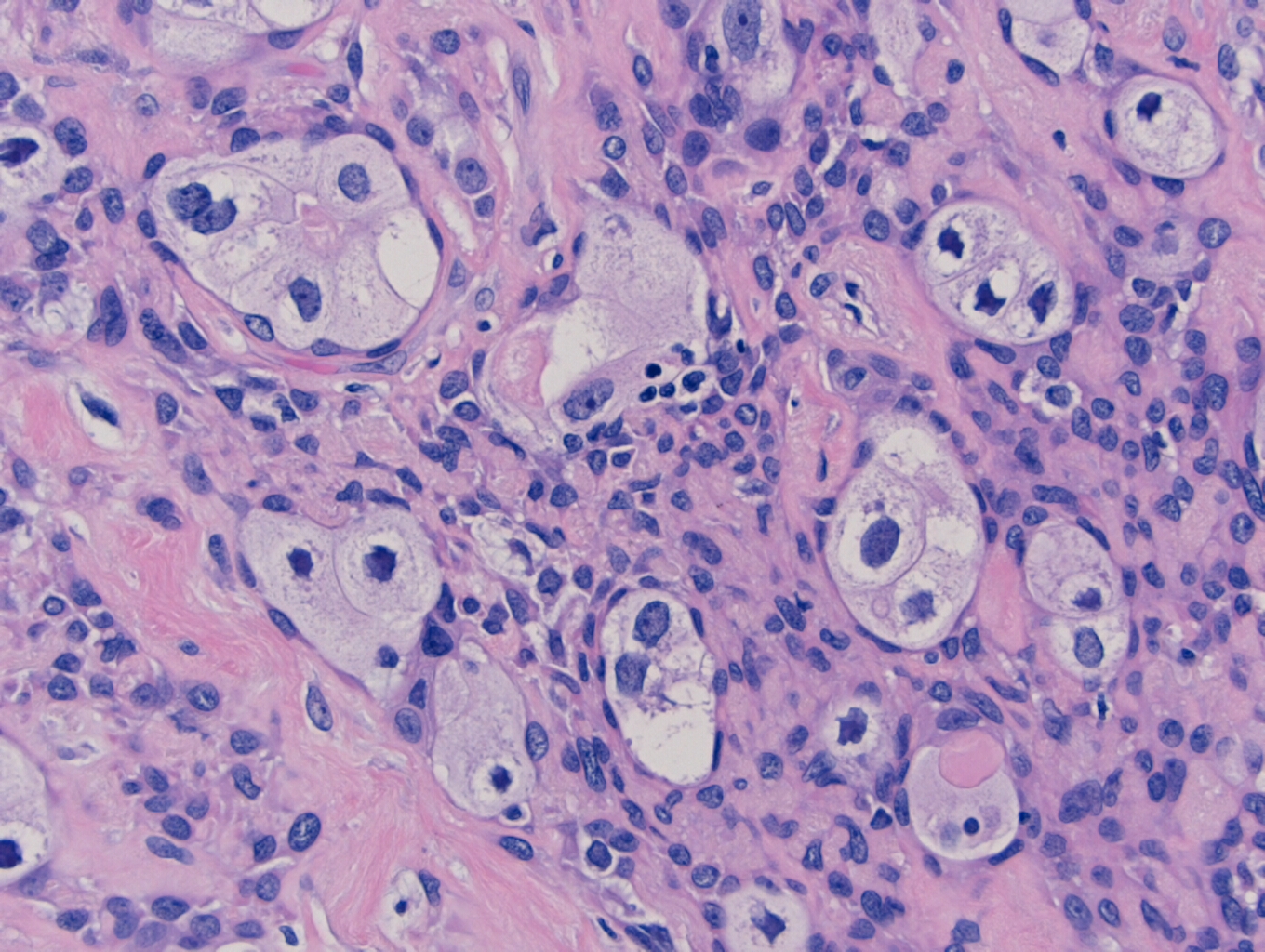
microscopy, the specimen contains nests of tumor cells ranging in size from 0.2
to 4.0 cm within a dense fibrous matrix. Although these deposits may represent
lymph node metastases, no residual lymphoid tissue is present. The tumor is
represented by residual pleomorphic adenoma and numerous soft tissue deposits
of pleomorphic adenoma (Figure 3). Admixed are broad areas of high grade
carcinoma with necrosis (Figure 4). Most regions show adenocarcinoma, although
a rare focus of squamous differentiation is also present. The lateral margin is
positive for carcinoma, and a pleomorphic adenoma component approaches within
0.1 cm of the medial margin. The anterior, posterior, inferior, and superior
margins are all free of tumor. No salivary gland tissue is identified.
In
addition, eleven frozen sections are submitted from various areas surrounding
the mass, with five of the eleven frozen sections demonstrating tumor deposits.
A right neck dissection is performed with following results:
Level
IB: 2 of 3 positive (largest deposit: 1.8 cm)
Level II
and III: 1 of 14 positive, Level II (1.9cm)
Level
IV: 1 of 8 positive (2.0 cm)
Based on
these results, the specimen was signed out as carcinoma ex-pleomorphic adenoma,
and designated as pT4aN2cMx
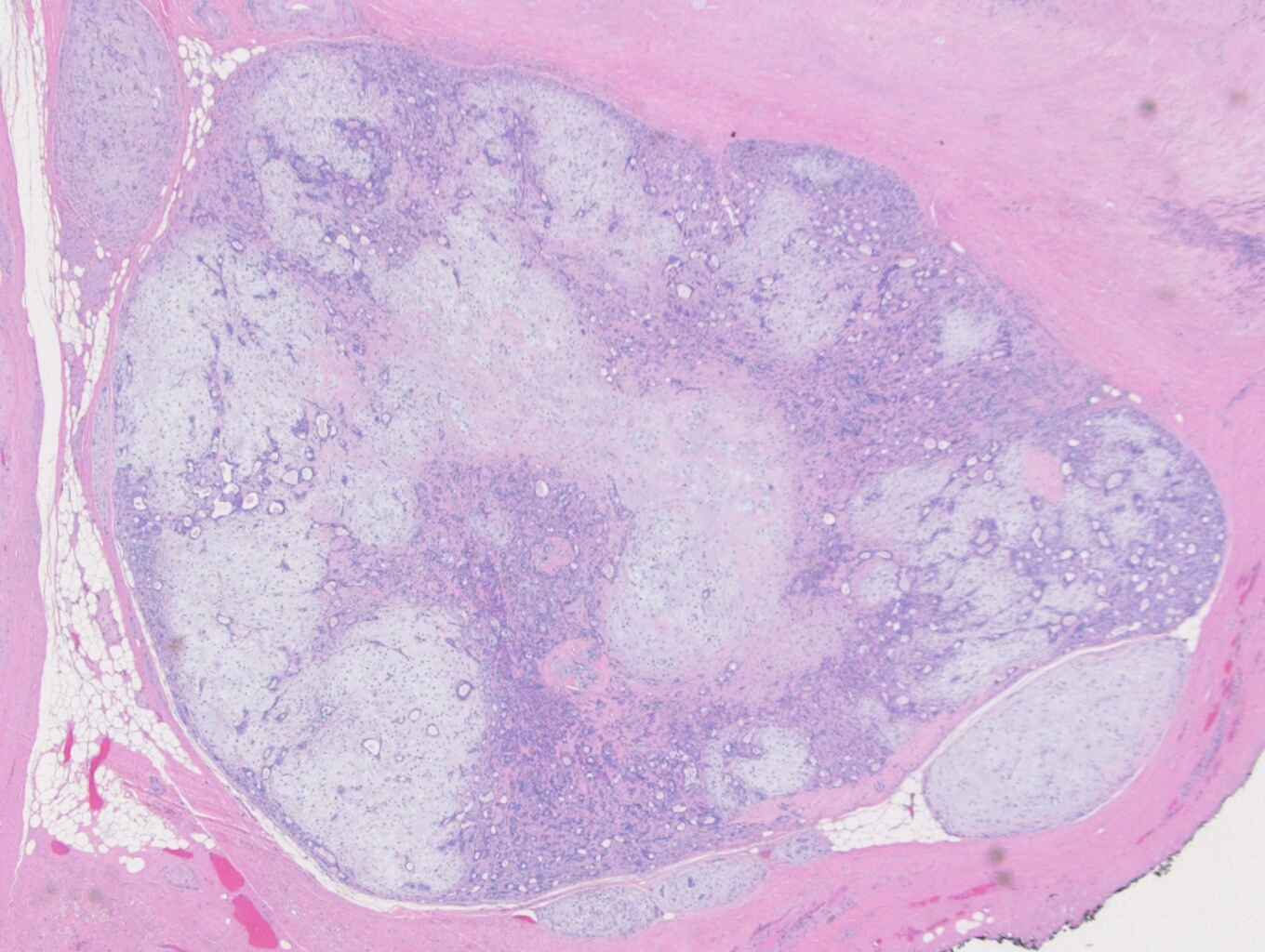
Figure 3. 2x photomicrograph showing a classic appearing pleomorphic adenoma with satellite nodules along the periphery
Discussion
Carcinoma ex pleomorphic adenoma
(CXPA) is a carcinoma that arises in a primary (de novo) or recurrent benign
pleomorphic adenoma (PA). While a PA is the most common salivary gland tumor,
accounting for approximately 80% of all benign salivary gland tumors, a CXPA is
quite uncommon, accounting for only 3.6% of all salivary gland tumors. CXPA is predominantly
found in the sixth to eighth decades of life, with a slight predilection for
females. CXPA arises most commonly in the salivary glands, in particular the
parotid and the submandibular glands. CXPA can also arise in the minor salivary
glands in the oral cavity, although these tumors tend to be smaller than their
counterparts in the parotid and submandibular gland. There have also been cases
of CXPA in the breast, lacrimal gland, trachea, and nasal cavity.
Clinically, CXPA presents as a firm, asymptomatic mass that can go undetected for years since they are not generally invasive. When the patient does experience any symptoms, with pain being the most common, it is usually due to the mass extending to adjacent structures. If the mass was to involve the facial nerve, paresis or palsy can occur. Other signs and symptoms include skin ulceration, mass enlargement, skin fixation, lymphadenopathy, dental pain, and dysphagia. The onset of symptoms can range anywhere from 1 month up to 60 years (such as with this case), with a mean onset of 9 years. Half of patients will have a painless mass for less than 1 year. Since these symptoms are similar to those of a benign PA, it’s important that the treating physician be aware of the possibility of a CXPA, especially considering the rarity of the cancer.
Grossly, CXPA appears as a firm,
ill-defined tumor, and can vary greatly depending on the predominant component.
If the PA is the predominant component, the mass may appear gray-blue and
translucent, and it could be possible to grossly differentiate between the PA
areas and the CXPA areas. If the malignant component predominates, then the
mass may contain cystic, hemorrhagic and necrotic areas.
Microscopically, CXPA is defined as having a mixture of a benign PA, admixed with carcinomatous components. Zbaren et al, in an analysis of 19 CXPA cases, found 21% of the tumors were composed of less than 33% carcinoma, 37% of the tumors were composed of 33-66% carcinoma, and 42% of the tumors were composed of greater than 66% carcinoma. Most often, the malignant component is adenocarcinoma, but can also include adenoid cystic carcinoma, mucoepidermoid carcinoma, salivary duct carcinoma, and other less common variations. In cases where the entire tumor is replaced by carcinoma, the diagnosis of CXPA will be based on the presence of a PA on the previous biopsy. Conversely, you could also have a tumor that is predominately composed of a PA, with sparse areas of malignant transformation, such as nuclear pleomorphism, atypical mitotic figures, hemorrhage and necrosis. The likelihood of malignant transformation increases with the length of the PA being present, from 1.5% at 5 years, up to 10% after 15 years.
CXPA can be further sub-divided into four categories based on the extent of invasion of the carcinomatous component outside the capsule: in-situ, non-invasive, minimally invasive, and invasive carcinoma.
#1) In-situ carcinoma occurs when nuclear pleomorphism and
atypical mitotic figures are found within the epithelial cells, but do not
extend out beyond the border of the myoepithelial cells (Figure 5).
#2) Non-invasive CXPA, which can include in-situ carcinoma,
is maintained within the fibrous capsule of the PA, but extends beyond the
confines of the myoepithelial cells. Non-invasive CXPA may begin to show
malignant transformation, but will overall behave like a benign PA.
#3) Minimally invasive CXPA is defined as <1.5 mm
extension into the extracapsular tissue, with a mix of benign PA components and
carcinomatous components.
#4) Invasive CXPA is defined as a > 1.5 mm extension into
the extracapsular tissue, and will begin to demonstrate more carcinomatous
components, such as hemorrhage and necrosis.
As the carcinomatous areas begin
to increase in prevalence, the PA nodules will begin to be composed of
hyalinized tissue with sparse, scattered ductal structures, and the malignant
cells will begin to decrease in size as they move away from the site of origin.
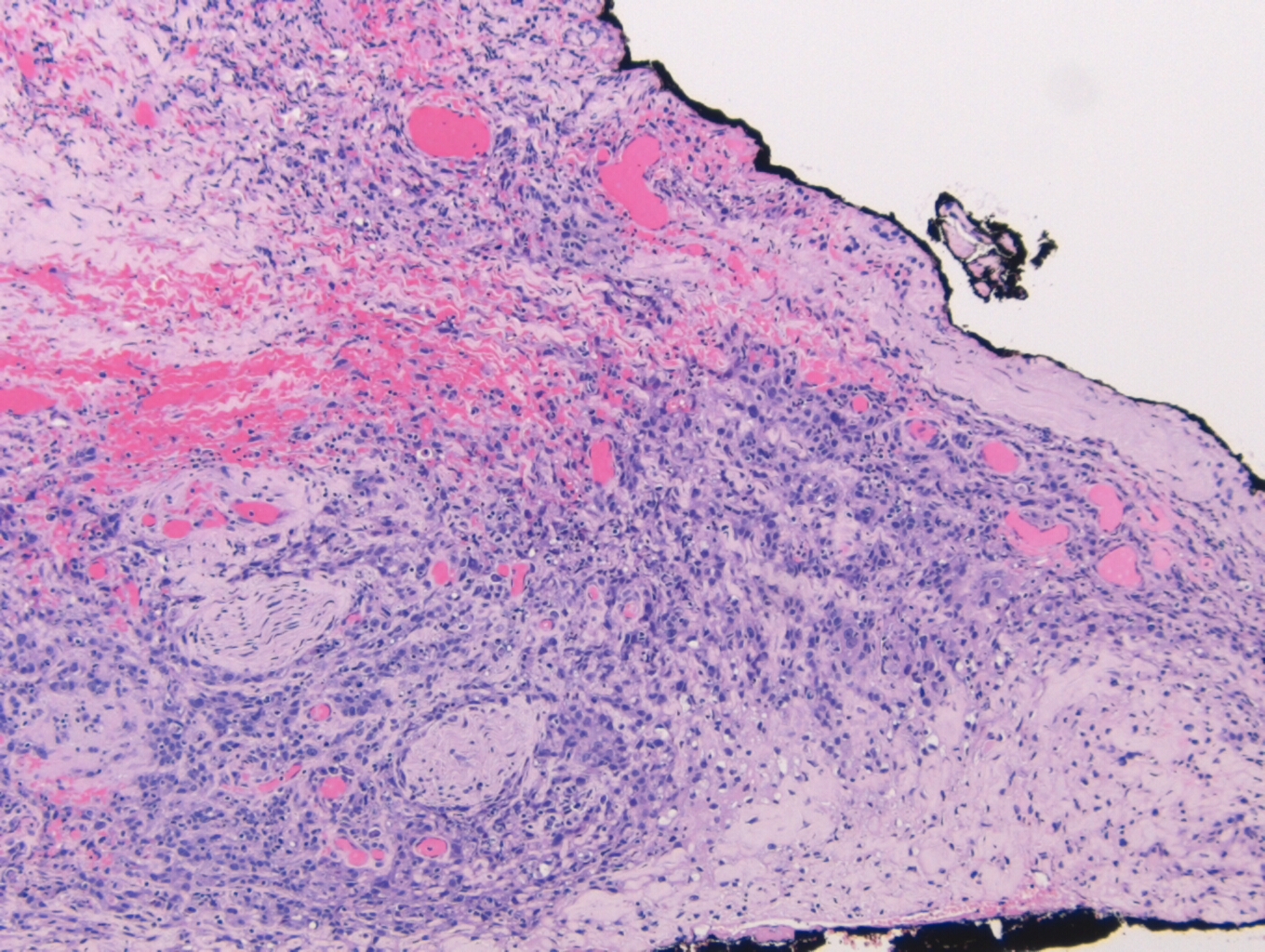
Perineural and vascular invasion can be easily identified as the tumor extends
into the neighboring tissue (Figure 6).
The development of CXPA has been
shown to follow a multi-step model of carcinogenesis with a loss of
heterozygosity at chromosomal arms 8q, followed by 12q, and finally 17p. Both
PA and CXPA demonstrate the same loss of heterozygosity, however, the carcinomatous
components exhibit a slightly higher loss of heterozygosity at 8q, and a
significantly higher loss of heterozygosity at 12q and 17q. The early
alterations of the chromosomal arm 8q in a PA often involves PLAG1 and MYC,
with the malignant transformation of the PA to a CXPA being associated with the
12q genes HMGA2 and MDM2.
Treatment for CXPA involves
surgery, radiotherapy and chemotherapy, with a parotidectomy being the most
common procedure performed. If a benign PA had originally been resected, but
residual remnants of the PA were left behind, then satellite PA nodules will
arise in its place (Figure 3). If in-situ, non-invasive or minimally invasive
carcinoma is suspected in the superficial lobe of the parotid gland, than a
superficial parotidectomy can be performed. Invasive carcinoma will result in a
total parotidectomy, with every attempt made to try and preserve the facial
nerve. If metastasis is suspected to the cervical lymph nodes, a neck
dissection may also be performed. Reconstructive surgery following the removal
of the tumor may be necessary, depending on where the tumor was resected from. Other
treatment options currently being considered include a combination therapy of trastuzumab
and capecitabine, as well as the possibility of a WT1 peptide based
immunotherapy.
Figure 5. 40x microphotograph demonstrating an in-situ carcinoma confined within the myoepithelial cells Figure 6. 10x photomicrograph of carcinoma at the lateral margin with areas of perineural invasion
References
Antony J, Gopalan V, Smith RA, Lam AK. Carcinoma ex pleomorphic adenoma: a comprehensive review of clinical, pathological and molecular data. Head Neck Pathol. 2011;6(1):1–9. doi:10.1007/s12105-011-0281-z
Chooback N, Shen Y, Jones M, et al. Carcinoma ex pleomorphic adenoma: case report and options for systemic therapy. Curr Oncol. 2017;24(3):e251–e254. doi:10.3747/co.24.3588
Di Palma S. Carcinoma ex pleomorphic adenoma, with particular emphasis on early lesions. Head Neck Pathol. 2013;7 Suppl 1(Suppl 1):S68–S76. doi:10.1007/s12105-013-0454-z
-Cory Nash is a board certified Pathologists’ Assistant, specializing in surgical and gross pathology. He currently works as a Pathologists’ Assistant at the University of Chicago Medical Center. His job involves the macroscopic examination, dissection and tissue submission of surgical specimens, ranging from biopsies to multi-organ resections. Cory has a special interest in head and neck pathology, as well as bone and soft tissue pathology. Cory can be followed on twitter at @iplaywithorgans.
Insulin antibodies are seen in two conditions: 1, in insulin-naïve type-1
diabetic patients, insulin antibodies are developed together with some other
autoantibodies against pancreatic islet cells; 2, in patients being treated
with insulin, antibodies can be developed against exogenous insulins, in both
type-1 and type-2 diabetes. These antibodies against exogenous insulins are
found in >95% of patients treated with porcine and bovine insulins (1).
Although the prevalence has decreased after the introduction of human insulin
and insulin analogues, it is still not uncommon to detect these antibodies in
insulin treated patients (2). However, these antibodies are rarely of clinical
significance and laboratory test for insulin antibodies in insulin-treated
patients has limited clinical value, except in rare cases where these
antibodies are found to have immunologic role, causing insulin resistance. In
some of these cases, postprandial hyperglycemia and nighttime hypoglycemia are
both described due to reversible binding of insulin from antibodies (3), and
patients were reported to respond to immunosuppressive therapies, and plasmapheresis
in severe cases.
We recently
worked up a case for possible immunologic insulin resistance caused by insulin
antibodies. In this case, patient is a 45 years old female with uncontrolled
type-1 diabetes. She was found to have all four antibodies positive, including
zinc transporter 8, islet
antigens glutamate decarboxylase 65 (GADA), IA-2A, and insulin antibodies. Patient
has been on multiple dose insulin injection (MDI) therapy, including insulin
determir, aspart and lispro. She was reported to be compliant with medications
and low carb diet. However, patient has poor glycemic control and presents with
recurrent diabetic ketoacidosis. She was given high doses of insulins, but
still presented with recurrent DKA and occasional hypoglycemia. Her HbA1c was
consistently at >10% with daily glucose measured up to 500 mg/dL.
Immunologic
insulin antibody and insulin receptor antibody were considered after ruling out
more common causes of her uncontrolled diabetes. These two tests were then performed
at a reference laboratory and patient was found to have positive insulin
antibodies to analog insulin (determir and lispro) and negative insulin
receptor antibodies. Significant insulin resistance by insulin antibodies was
not found and the antibodies level did not suggest immunosuppressive therapy. Still,
given her poor controlled diabetes, patient’s insulin was switched to human
insulin and she was also recommend for pancreas transplant.
Greenfield JR, Tuthill A, Soos
MA, Semple RK, Halsall DJ, Chaudhry A, O’Rahilly S. Severe insulin
resistance due to anti-insulin antibodies: response to plasma exchange and
immunosuppressive therapy. Diabet Med. 2009 Jan;26(1):79-82. doi:
10.1111/j.1464-5491.2008.02621.x.
Hall TR, Thomas JW, Padoa CJ, Torn
C, Landin-Olsson M, Ortqvist E, Hampe CS. Longitudinal epitope analysis of
insulin-binding antibodies in type 1 diabetes. Clin Exp Immunol. 2006
Oct;146(1):9-14.
Hao JB, Imam S, Dar
P, Alfonso-Jaume M, Elnagar N, Jaume JC. Extreme Insulin
Resistance From Insulin Antibodies (Not Insulin Receptor Antibodies)
Successfully Treated With Combination Immunosuppressive Therapy. Diabetes
Care. 2017 Feb;40(2):e19-e20. doi: 10.2337/dc16-1975. Epub 2016
Dec 1.
-Xin Yi, PhD, DABCC, FACB, is a board-certified clinical chemist, currently serving as the Co-director of Clinical Chemistry at Houston Methodist Hospital in Houston, TX and an Assistant Professor of Clinical Pathology and Laboratory Medicine at Weill Cornell Medical College.
In today’s
hematology lab, when physicians order a CBC with differential, they typically
request a CBC with automated differential. Thus, up to 85% of our CBCs are
autovalidated because they are entirely within normal range, with no instrument
flags. This leaves the technologist time to spend on those slides that do need
a manual review. In reviewing a slide, we evaluate the WBCs, RBCs and
platelets, and must pay attention to the counts as well as morphology.
But, what do we
do when we have a cell we cannot identify? When we perform a manual
differential under the microscope, technologists will joke or tell stories
about the legendary “skipocyte”; that cell which, while it does not look malignant
or clinically significant, we still can’t decide what it is, so it’s skipped.
Perhaps the best way to deal with these cells would be to get consensus from
other techs or the Hematology supervisor or to request a pathology review.
However, despite the fact that we are taught that there is no such thing as a
skipocyte, there are times when a tech will ignore the cell, hoping they don’t
see another one. But, what do we do when we see smudge cells? Are they
skipocytes? What exactly are they? Do we ignore these? Are they clinically
significant? Do we count them as their own category of cells? Or something
else?
Firstly, what is a smudge cell? Smudge cells, or basket cells,
are remnants of leukocytes. They have no cytoplasm, and sometimes all that can
be seen are smashed nuclei. Smudge cells are formed from leukocytes, typically
lymphocytes, that are fragile, and are destroyed or smudged in the physical
process of making a smear. But, what if the instrument makes the smear? In
recent years, more labs are using automated analyzers that prepare and stain
blood smears. Even though these have instrument settings based on the physical
characteristics of each sample, we still tend to observe these traumatic
injuries to leukocytes with automated slide making. Whether we make slides
manually or the instrument makes them, these fragile cells appear on the
stained slide as ruptured cells called smudge cells.
Image 1. Smudge cells seen on peripheral blood smear.
Smudge cells
have also been called Gumprecht shadows, named after German scientists and
researcher Ferdinand Adolph
Gumprecht, who observed these on slides of patients with chronic
lymphocytic leukemia (CLL). Smudge cells in patients with CLL are ruptured
B-cells, but they can’t be distinguished morphologically from other
disintegrated lymphocytes. We also see leukocytosis and smudge cells in viral conditions
and chronic inflammatory diseases. However, the term Gumprecht shadows is
reserved only for smudge cells in CLL cases.
Knowing what a
smudge cell is, how do we handle them? Do we report the presence only? Do we
count them? Or, do we ignore them entirely? Smudge cells are not skipocytes!
For many years smudge cells were considered to be simply artifacts of slide
making. More recently, studies have been conducted that show that there may be clinical
significance to the number of smudge cells seen. While smudge cells are not
diagnostic of CLL, it has been shown that, in newly diagnosed CLL, a larger
percentage of smudge cells is a better prognostic factor. Patients with >30%
smudge cells show longer times before requiring treatment and longer survival
rates than patients with fewer smudge cells. These studies focused on vimentin,
a protein that is important in lymphocyte cellular rigidity. Patients with low
vimentin have more smudge cells and better survival rates.1,2
If we are
performing a slide review, we are reviewing these slides because of some sort
of instrument flag or rule trigger. There are several theories as to how smudge
cells can be handled, and studies have been done to compare these theories.3
Laboratories have SOPs in place to guide technologist review and
reporting, yet, I have noticed considerable variation in handling of smudge
cells both within our lab and between labs. These pesky artefacts can be puzzling in
both traditional (under the microscope) and digitized (CellaVision) microscopy
and new technologists or unfamiliar operators can easily be misled.
If we perform our manual differentials traditionally, under
the microscope, we will no doubt notice the presence of smudge cells. It is
important not to pass by these or consider them skipocytes. Some labs count
these as their own category of cell and some labs merely report the presence of
smudge cells. Other labs do not report smudge cells at all, with the exception
being in known cases of CLL. In these CLL differentials, if the WBC count is
very high, it may also be recommended to do a 200 cell differential. But, what
happens when the manual diff doesn’t match the automated diff? The hematology
analyzer will accurately count fragile cells, still intact in the specimen, and
include them in the differential. If the cells then disintegrate on smear
making, we see smudge cells on the slide. If we do not count these, this can
affect the percentage of cell types in the differential, and potentially, in a
patient with a low WBC, affect the absolute neutrophil count (ANC). If we are
performing the manual differential (diff) in CellaVision, CellaVision
identifies smudge cells and puts them in a separate category, but these are not
reported as part of the diff. These are a ‘heads up’ to the technologist that further
steps need to be taken to report out a differential. The importance of
recognizing smudge cells is illustrated in Table 1 below for a patient sample
with WBC 3.6 x 103/μL.
Table 1. Numbers of cells counted in three differentials on sample with WBC 3.6 x 103/μL.
The automated differential
(auto diff) in this example, with 12% neutrophils counted, has an absolute
neutrophil count of 432/ μL,
which is considered critical (critical <500/μL). Fragile lymphocytes are intact in the blood
sample and are counted by hematology analyzers.
The 200 cell
manual differential above merely notes the presence of smudge cells, but no
quantifier is given. The ANC here is not critical (774/μL) and the lymph% is only 61, possibly leaving the
physician to question how many smudge cells were present, and what the true
lymph% may be.
In the CellavVision
differential in Table 1, based on 100 WBCs counted, the total percentages of
Neuts is 20%, lymphs 61% and monos 19%. If an unexperienced tech did not notice
or investigate the 68 smudge cells, the manual differential (manual diff) reported
from the CellaVision would be very different from the auto diff, and has an ANC
of 720/μL, above the
critical range.
If however, the
smudge cells in CellaVision were reported as a separate category, our
differential would now be based on 168 cells counted. 100 WBCs counted plus the
68 smudge cells counted = total of 168 cells counted. Our neut% is now 11.9 (20/168*100),
lymph % 36.3(61/168*100), monos% 11.3 (19/168*100) and smudge cell % 40.5 (68/168*100).
This ANC matches that from the auto diff. And, if we further consider that the
smudge cells are lymphocytes, this brings the count to 11.9% neuts, 77% lymphs
and 11.3% monos. (68 +61 = 129/168*100 = 77% lymphs) which closely matches our
automated differential.
Lastly, the ‘something else’, is that we
can make an albumin smear on these specimens. It has been a practice in labs to
perform a manual differential on an albuminized blood smear when a certain
number, defined by SOPs, of smudge cells are seen. If this is your lab
procedure, it is important to recognize the presence of smudge cells on the
manual differential or CellaVision differential and take the steps to make an
albumin smear. Adding a drop of albumin to a few drops of the patient blood can
add protein to the specimen and prevent the formation of smudge cells. Table 2
shows the manual diff on the sample in Table 1, performed on the albuminized
slide. Note that this eliminates the smudge cells and corrects the diff results
to match the original automated differential.
Table 2. Albumin smear results on sample from table 1.
It can be seen from these examples, that
the method of counting differentials with smudge cells can alter the results
reported to the physician. Any of the differential methods above that count
smudge cells give essentially the same results. If
smudge cells are not counted, the lymphs will be under reported and the
neutrophils will be over represented compared to the auto diff. Excluding
smudge cells from the manual differential count or merely reporting their
presence without quantification will also yield unreliable results, and then necessitates
performing an albumin differential.
If
we are to choose between an albumin differential and an automated differential,
which studies have shown to be equivalent3, making and staining an
additional smear is time consuming and can affect turnaround times. Thus, guidelines have been suggested
that the first choice for handling pesky smudge cells is to review the smear
and report the automated diff with a morphology comment that smudge cells are
present. If automated diffs are not available, smudge cells should be counted
as lymphocytes, or in a separate category4, as illustrated in Table
1. Study findings indicate that this method is sufficient for reporting a
reliable manual differential on known CLL patients3. By counting
smudge cells separately, however, as discussed previously, these numbers can be
used in newly diagnosed cases of CLL as a prognostic indicator.
There is still debate on the value of reporting smudge cells on
routine CBC smears. In most routine cases, an auto diff without quantitating
smudge cells is considered sufficient. Pathologists, however, differ on whether
smudge cells should be reported.
The best course of action is always to consistently follow
your own lab’s SOPs, to be aware of flags, rules triggered and operator alerts
with regard to smears, and to always be on the lookout for smudge cells. They
are not skipocytes!
References
Nowakowski
GS, Hoyer JD, Shanafelt TD, et al. Percentage of smudge cells on routine blood
smear predicts survival in chronic lymphocytic leukemia. J Clin Oncol.
2009;27(11):1844-1849.
Amal
Abd El Hamid Mohamed, Nesma Ahmed Safwat. New insights into smudge cell
percentage in chronic lymphocytic Leukemia: A novel prognostic indicator of
disease burden. The Egyptian Journal of
Medical Human Genetics,
19 (2018) 409–415
Gene
Gulati, Vandi Ly, Guldeep Uppal, Jerald Gong, Feasibility of Counting Smudge
Cells as Lymphocytes in Differential Leukocyte Counts Performed on Blood Smears
of Patients With Established or Suspected Chronic Lymphocytic Leukemia/Small
Lymphocytic Lymphoma, Laboratory Medicine, Volume 48, Issue 2, May
2017, Pages 137–147, https://doi.org/10.1093/labmed/lmx002
Denis
Macdonald, MD, MBA, FRCPC, FCAP; Harold Richardson,et al. Practice Guidelines
on the Reporting of Smudge Cells in the White Blood Cell Differential Count. Arch Pathol Lab Med—Vol 127, January
2003
Luci
Maria Sant’Ana Dusse; Tamiris Paula Silva, et al. Gumprecht shadows: when to
use this terminology? J Bras Patol Med
Lab, v. 49, n. 5, p. 320-323, 2013
-Becky Socha, MS, MLS(ASCP)CM BB CM graduated from Merrimack College in N. Andover, Massachusetts with a BS in Medical Technology and completed her MS in Clinical Laboratory Sciences at the University of Massachusetts, Lowell. She has worked as a Medical Technologist for over 30 years. She’s worked in all areas of the clinical laboratory, but has a special interest in Hematology and Blood Banking. When she’s not busy being a mad scientist, she can be found outside riding her bicycle.
Last
month we discussed the rules and requirements for how to properly perform
proficiency testing (PT) within your laboratory. In part 2 of this 3-part
series we’ll review the rules associated with evaluating your results, and how
to investigate any unsuccessful surveys. Still to come in part 3 we will look
into how to utilize your PT results to monitor for trends and shifts in your
values.
The
rules:
Performance Review: Laboratories must initiate and document a review of their PT performance evaluations within 2 weeks of notification that results are available. This includes a review of both graded and non-graded/educational analytes and events as well.
Key
things to note: Even though educational samples are not formally graded, you
should still verify the accuracy of your results, with appropriate follow-up
for any failures. CAP specifically requires you to evaluate these educational
challenges as well. Whether the sample is graded or not does not change the
fact that you had an incorrect result.
Unsatisfactory Performance: For any unsatisfactory results,
you are required to perform a root cause analysis to determine why (see below
for guidance). This also includes any clerical errors – you need to evaluate
your process and find ways to prevent these simple errors from happening again.
If they are happening with PT samples, it is possible they are happening with
patient samples as well.
Cessation of Patient Testing: Unsatisfactory events indicate
that there was a problem with that particular survey; whereas unsuccessful events indicate
there has been a pattern of unsatisfactory events/samples and a larger problem
exists. If a pattern of poor performance is detected, you may be asked by your
local state department of health to cease all testing for a particular analyte.
Key
things to note: This also applies to clerical errors. Even if there was no
technical problem with the accuracy of your results, failure to submit results
on time or clerical errors made while submitting can also have severe impacts
on your ability to continue offering that test.
Remedial Action: If you’ve been notified by your
PT provider or state DOH to cease testing, there are extensive steps that must be
completed to prove that the problem was correctly identified and corrected. You
must also identify where samples will be referred to for tests you are unable
to perform in-house.
Key
things to note: If testing has been removed from your laboratory, you will be
required to demonstrate successful performance in 2 consecutive PT survey
events for the analyte(s) in question before being granted permission to resume
patient testing. This can cause significant delays and financial impact for
your organization.
Root Cause Analysis: Investigate to determine who,
what, why, when, and how the event occurred. Be sure to evaluate all phases of
testing to ensure you identify all potential causes.
Pre-Examination:
Human Resources – evaluate the training and competency records for staff involved in the handling and testing of samples.
Facilities – reagent inventory control & storage temperatures, equipment maintenance and function checks
Standard Operating Procedures (SOPs) – staff compliance with written policies, bench excerpts are current and valid, document version control up to date
Specimen –test requisition/order entry (was the correct test code ordered/performed?), labeling (were aliquot/pour off tubes properly labeled?), transport (was appropriate temperature requirements maintained until testing performed), quality (was there visible deterioration with the sample prior to testing or cracked/damaged tubes received?), quantity (was the original sample spilled or leaking causing an incomplete aspiration of sample by your instrument?)
Examination:
Method
Validations – were instruments current with calibration requirements, any bias
noted during instrument correlation studies, values being reported within the
verified AMR
Environmental
Controls – temperatures/humidity within tolerance limits, for light sensitive
studies (bilirubin) was there excessive exposure of the samples to light prior
to testing, excessive vibrations occurring that may have affected results
(nearby construction or a running centrifuge on a shared work bench)
Quality
Control – did QC pass on the day of testing, was QC trending or shifts noted
that month
Analytical
Records (worksheets) – were sample results transcribed correctly between the
analyzer and worksheet, between the worksheet and LIS
Instrument
Errors – were any corrective actions or problems noted for the days before,
during, or immediately after testing of PT occurred
Testing
Delay, Testing Errors – were samples prepared and not tested immediately
leaving them exposed to light or air which may affect results (blood gas
samples), any errors or problems noted during testing that may have caused a
delay or affected accuracy of results
Post-Examination:
Data
& Results Review – check for clerical errors, was data trasmitted correctly
from the instrument into LIS, was data entered correctly on your PT provider
entry submission forms
Verification
of Transmission – did your results correctly upload to the PT provider website,
was there an error or failure with submission
Review
of LIS – are your autoverification rules set up correctly, is the
autoverification validation current with no known issues
Patient
Impact – perhaps
the most important step to take when reviewing PT failures, you need to
determine what impact your failure had on your patient results. Depending upon
the identified root cause and how different your values were from the intended
response, this can potentially pose a severe impact on your patient values
tested at the same time as the PT samples.
Involve
your medical director to determine if the discrepancy in results is clinically
significant. Perform a patient look-back to review patient values for the same
analyte with the failure during the time period in question. Evaluate the bias
that was present, and if deemed to be clinically significant then corrected
patient reports will need to be issued with a letter from the medical director
explaining why. If it was decided that the discrepancy is not clinically
significant, document this in writing and keep on record with your complete
investigation response.
Corrective Actions/Preventative
Actions– use the following set of questions to help guide you
in ensuring that the problem identified during your root cause analysis will
not occur again:
What
changes to policies, procedures, and/or processes will you implement to ensure
there will not be a repeat of this problem?
Do
any processes need to be simplified or standardized?
Is
additional training or competency assessment needed? If so, identify specific
team members to be trained, and who will be accountable for performing and
documenting this training.
Is
additional supervisory oversight needed for a particular area or step?
Are
current staffing levels adequate to handle testing volumes?
Would
revision or additional verification of the LIS rules address or prevent this
problem?
How
can the communication between laboratory, nursing, and medical staff be
improved to reduce errors in the future?
Continuous Process
Improvement –
after identifying the true root cause(s) for the failure and implementing corrective/preventative
actions, you need to evaluate the effectiveness of those improvements. Have
they been sustained? Are they working to correct the original problem? Have you
created new problems by changing the previous process?
Quality
Management Meetings – if necessary, increase the frequency of these meetings
during the evaluation period for timely feedback to management and staff
Implement
internal audits and quality indicators to check for potential issues
Access
the specimen transport conditions to ensure they meet test requirements
Evaluate
and monitor your turnaround time metrics to track problem specimens and impact
of testing delays
If
necessary, increase the frequency when QC is performed or calibration frequency
if stability issues are identified
Performing
a thorough root cause analysis for any failures will allow you to implement
appropriate corrective actions that will address the true issues. Having a
robust quality management program will help ensure these issues are identified
and corrected in a timely manner, and reduce the potential for the dreaded
Cessation of Patient Testing letter from your local DOH.
Coming
up in the final installment of this series on PT testing, we’ll review all of
the quality indicators and data that can be found in your PT evaluation reports
to help ensure you’re on track for accurate patient values.
-Kyle Nevins, MS, MLS(ASCP)CM is one of ASCP’s 2018
Top 5 in the 40 Under Forty recognition program. She has worked in the
medical laboratory profession for over 18 years. In her current
position, she transitions between performing laboratory audits across
the entire Northwell Health System on Long Island, NY, consulting for
at-risk laboratories outside of Northwell Health, bringing laboratories
up to regulatory standards, and acting as supervisor and mentor in labs
with management gaps.