Large biological and chemical spills are not a common
occurrence in the laboratory. That’s a good thing, but when they do occur, they
can create a very dangerous situation. It is vital that lab staff know how to handle
such events even though they may not be commonplace.
Some laboratories differentiate between large and small
spills. They may have an emergency number to call for a hazardous spill
response team. Other smaller facilities simply don’t have that in place. Either
way, it’s important for laboratory professionals to know they are the experts
about the biological and chemical materials they use, and they need to be in
charge as the experts when a spill situation needs to be managed.
Most laboratory spills can be managed using a standardized step-wise
process known as the S.P.I.L.L.E.D. procedure. I don’t usually ask lab staff to
memorize the acronym, but having the information contained on a poster with the
lab spill kits can make a clean-up procedure go smoothly.
S = Secure the Site – Make sure no one walks through the
area where a spill has occurred. It could be a dangerous situation if a
hazardous chemical is spilled, and you would never want someone slipping in the
area or tracking the spilled material to another area.
P = Protect Yourself – Arm yourself with the
appropriate Personal Protective Equipment (PPE). In a lab spill event, this
would mean using a lab coat, gloves, and face protection to prevent accidental splashes.
I = Inspect the Spill – Look to see what was spilled.
If it is a hazardous chemical, is there a concern about fumes? Obtain a Safety
Data Sheet to see if section 6 will give any special information about handling
the accidental release or spill of that chemical. Consider other spill concerns
such as broken glass or possible ignition sources if flammable material is
involved.
L = Lay Down a Barrier – If the spill is large and
spreading, lay down spill pillows or booms designed to contain a flow of
liquids. Surround the spill area with these materials. Sometimes, the use of an
emergency shower can create the need for a barrier to be made.
L = Lay Down Absorbents – No matter the size of the spill,
the next step is to place any absorbent powders, granules or clean-up pads to
soak up the spilled material. If the absorbent is also a neutralizer, make sure
you allow the necessary time for neutralization to occur.
E = Extract the Mess – Use implements to pick up the
materials used for stopping and absorbing the spill.
D = Dispose of the Waste – Properly dispose of all
materials involved with the spill clean-up. If there was glass involved, be sure
to use a sharps container. Biohazard
material should go into an appropriate container, and chemical waste materials
may need to be disposed of separately for pick-up by a chemical waste vendor.
Lab staff should be able to access spill control materials
quickly, and the necessary items should be stored in a location designated by
signage. Perform an inventory of spill supplies and make sure there are
adequate materials that could handle spills of the biohazards and chemicals stored
and used in the department. Be sure items in the spill kit are not expired, and
if there is no expiration date for absorbent powders, check them at least
annually for effectiveness.
All laboratory staff need to have complete spill clean-up
training. Give information about the types and locations of spill kits and how
to handle various types of spills that can occur. Once that training is done,
it will become important to perform spill drills in the department. Drills can
be performed a number of different ways, but a common method involves having a “victim”
spill water onto the floor and claim the material splashed into their eyes.
Watch from a distance to see how the staff reacts. Do they provide appropriate
first aid? Do they inspect the container label? Do they access the correct
clean-up supplies and facilitate cleaning efficiently? Make notes of how the
drill went, discuss them with the staff, and repeat the drills until all staff
are comfortable with a spill situation.
Biological
and chemical spills should not be a common occurrence in the lab. When they do
occur, however, the situation can become serious quickly, and a fast and effective
clean-up needs to occur. Because these events are rare, it becomes important to
provide regular spill training and drills so staff can remain ever-ready to
handle them.
–Dan Scungio, MT(ASCP), SLS, CQA (ASQ) has over 25 years
experience as a certified medical technologist. Today he is the
Laboratory Safety Officer for Sentara Healthcare, a system of seven
hospitals and over 20 laboratories and draw sites in the Tidewater area
of Virginia. He is also known as Dan the Lab Safety Man, a lab safety consultant, educator, and trainer.
I don’t think anyone enjoys filling out
the paperwork at a doctor’s office. For transgender individuals, this can be an
experience that ranges from irksome to offensive. Most intake forms don’t allow
for expression of their gender identity. Furthermore, confusion on gender and
sex can create real confusion and healthcare failures in several places that
laboratory medicine encounters a transgender individual.
Arguably the first place the lab
encounters a transgender patient is via the phlebotomist. These professional
collectors of blood must confirm two patient identifiers, which are often name
and date of birth. The “name” used is the legal name. Using a transgender
person’s “dead name” (name given at birth) represents a gender they do not want
to be associated with and can be a very offensive experience. “Isn’t it obvious
that name is not what I look like?”
While names can be legally changed, this
happens with varying difficulty and legal cost in different states. A solution
is to improve training of phlebotomists to explain the necessity of confirming
a legal name so lab results are properly matched to the patient. Additionally,
front-desk intake workers should be similarly trained to interact with
transgender patients when recording demographic information. This can be aided
by electronic health records (EHR) becoming more flexible and inclusive of the
gender diversity.
Traditionally, EHR would only include one
field for SEX: M or F.
Several in the laboratory community have
asked how many different gender options should be included? Facebook included
up to 71 options in 2017. That’s a big step up from the 2 traditional EHRs are
built around.
The World Professional Association for
Transgender Health (WPATH) executive committee in 2011 outlined the recommended
fields to include in EHR: preferred name, sex assigned at birth, gender, and
pronoun preference. EHRs are evolving and can be flexible depending on the user
requirements. At my program, we use EPIC at 3 different different sites (children’s,
county and university hospitals) and each has a different version.
From what I’ve seen preferred name is an easy addition and would not interfere with
functions of the EHR or Laboratory Information Systems (LIS), which is the
Lab’s version of EHR.
If the field for sex assigned at birth is different from gender, then it would clear up any confusion about whether the
person is transgender and then they should be addressed by the pronouns
matching the gender. While there is a spectrum of genders, only transgender males
and transgender females are of a high enough prevalence to have medically
relevant recommendations. Plus, if a system at least starts here, they could
expand further as necessitated by their population.
EHR could include preferred pronouns, but I haven’t seen this implemented in an EHR
yet. Ideally, you would just use the pronouns that match the intended
appearance of the individual (ma’am to someone wearing a dress, etc.).
Lastly, I think Legal sex should be added to the EHR as well. One of our hospitals
has this and it makes several processes easier such as processing hormone
medication.
Legal (or administrative) sex, sex assigned at birth, and gender data fields provide the clearest and simplest picture of a patient
and should be a minimum for labs making recommendations for changes to HER.
Next month I will describe in greater
detail the issues that can arise in the lab when gender or sex are entered
incorrectly in the system for transgender patients and how this can negatively
affect care delivery.
References
Deutsch MB, Green J, Keatley J, Mayer G, Hastings J, Hall AM, World Professional Association for Transgender Health EMR Working Group. Electronic medical records and the transgender patient: recommendations from the World Professional Association for Transgender Health EMR Working Group. J Am Med Inform Assoc. 2013 Jul-Aug; 20(4):700-3.
Gupta S, Imborek KL, Krasowski MD. Challenges in Transgender Healthcare: The Pathology Perspective. Lab Med. 2016 Aug; 47(3):180-188.
-Jeff SoRelle, MD is a Chief Resident of Pathology at the University of Texas Southwestern Medical Center in Dallas, TX. His clinical research interests include understanding how the lab intersects with transgender healthcare and improving genetic variant interpretation.
I merrily wait in line at Starbucks for my iced
cappuccino with soy milk, pay $5+ for $0.25 worth of goods poured into my
$14.00 souvenir mug, and walk out the door with my head held high, joyous with
the privileges of conspicuous consumption. My server was super-cheery and the
brief exchange we had was so pleasant—they really love me! I need that high because I am off to the
Department of Motor Vehicles (DMV) for a driving-related task and know–just
know–that there will be an incredibly long line at the end of which sits a
disgruntled government employee who doesn’t care if I show up or not. Their
motivation to help us is non-existent. “Why would anyone ever work here?” I
ask, sipping my delicious beverage.
Today, a doctor called someone in the United
States (US) and told them the biopsy taken from their leg earlier this week has
come back as invasive cancer. A bit distraught and nervous, the patient called
up a nationally recognized cancer center, from which they only live a few
miles, and on the end of the line is a caring, pleasant voice who informs them they
can be seen today! The valet parking is gorgeous, the building is gleaming with
glass and steel, and every face they see as they journey from check-in to
clinic is smiling, compassionate, and sincere. Their nurse and then doctor are
both genuine people with their patient’s best interest in mind, and they
carefully and completely explain what has been found, what needs to be done, and
how they are going to get through all of this together. As they depart, the
receptionist grabs them for a brief moment to return their private insurance
card and waves at them as they depart, adding, “We will see you soon!”
Today, someone in Africa went back to the hospital—an
8-hour journey from their home—where their biopsy was performed a month ago,
hoping to get the result. After several people searched multiple offices and
inquired with several people, the result is found and brought to them, a single
piece of paper. Payment is required before they can receive the biopsy results.
They have brought money with them, which they gathered from three neighbors, their
brother, and by selling some chickens, and pays for the report. They read the
report and, at the bottom, notices that it says additional testing is needed.
Confused, they ask for help and a pathologist comes to find them. Respectfully,
the pathologist explains that additional testing is needed, which is not
available in the hospital despite the pathologist’s strong desire to have it,
but they can send the biopsy to a lab elsewhere to do the testing which will
cost about 3 times what they just paid for the primary report. They happen to
have enough and pay the amount requested. The report will be back in about a
month. Two months later, they have returned to the hospital for the 4th time
and the report is now available. The testing that was done simply confirms that
the primary diagnosis is accurate. They go to the oncology clinic on the same
campus and sit in the waiting area with 3 dozen other people. They sleep at the
clinic overnight outside with about a dozen people. The following afternoon, they
are finally seen and the oncologist reviews the report. He notes that if the
patient had come to the clinic as soon as they had the biopsy result three
months ago, a simple surgery would have cured them of this lesion. But now,
because they waited so long, there is only chemotherapy available which is
expensive and, the oncologist reports, doesn’t actually work very well for this
tumor.
Before you shed a tear for this terrible situation
(while I sip my cappuccino and a nurse begins someone’s chemotherapy in a
shiny, brightly lit, and expansively windowed infusion unit not far away), we
have to ask ourselves what is really going on? First and foremost, this is an
allegory to make a few points but the situation is repeated over and over again
every day in the US and Africa. However, as a simple, superficial explanation,
the person with cancer in the US is receiving their cancer therapy from
Starbucks and the person in Africa had to go to the DMV.
Cancer care in the United States is almost
entirely in the private sector, dispersed among the 1500 cancer treatment
facilities, of which 70 are comprehensive cancer centers.[i]
Based on the US population, the expected cancer rate, 100% detection, and 240
working days for a given cancer center, there are on average only 5 new
patients per day per cancer center. Is that why one can often get that appointment
right away in a major cancer center or is it really a concierge customer
service effort? A standard private insurance plan for which I pay, for example,
$250 per month and my employer pays $1300 per month is accepted by cancer
centers and results in small co-pays for multiple appointments, which can be
covered with a Flexible Spending Account (FSA) or Health Savings Account (HSA).
On insurance statements after appointments, some of the services received cost
thousands of dollars but the patient portion was only, say, a hundred dollars,
again, which may be paid with FSA/HSA. It’s so great that we have insurance
because the insurance company is bearing the brunt of costs. But are they?
In the United States, 79% of facilities providing
health care are private, a mix of non-profit and for-profit.[ii]
But 64% of all healthcare in the United States is paid for by the US government
through Medicare, Medicaid, the Veterans Administration (VA) system, and Children’s
Health Insurance Program (CHIP).[iii],[iv]Since
almost every cancer care facility is private (or, stated another way, “not
free”), that means that for every one of us at the cancer center getting
treatment, for which we and our employer are paying through insurance, there
are two people getting the same treatment at the same high-level quality of
care for which the government is paying. Those other deductions from our
paychecks for Medicare and Medicaid (which everyone pays, regardless of how
old, as long as they are employed and regardless of their own health insurance
plan) are going towards the 64% coverage. The point is not that the US
healthcare system is expensive. The
point is that there is a lot of revenue and resource being put into the
healthcare system and, thus, there is a high-quality product or experience that
is available.
If we look at any low GINI index country and
compare their GDP with the US GPD and compare their spending on healthcare as a
% of GDP, we don’t even need to do the math to see that there is very little
money per person available in the system for any type of healthcare. The
challenge in low-resourced settings (by which it is meant low-resourced
patients in low-resources locations) is both a lack of funding available to
provide healthcare services along with a lack of “stuff” to provide those
services. We can invoke the law of supply and demand to try and argue that the
people can rise up and demand more healthcare facilities and “someone” will
meet that supply. In the US, this results in the Starbucks model. In a
low-resourced setting who has the incentive to meet that supply? Where does the
government get the money from to create such a system? What private corporation
is going to start a healthcare program that provides universal coverage
regardless of what you can pay?
The answer is really quite simple. This model of
healthcare is insufficient for cancer and isn’t going to work for all patients.
Moreover, the Starbucks model is not really applicable, sustainable, nor
equitable. When we go to Starbucks for their coffee, to some degree, our choice
of Starbucks is because of the a) flavor of the coffee, b) cost of the coffee,
c) perception of the coffee, and/or d) convenience of the coffee. We could
always choose Dunkin’, Peet’s, Tim Horton’s (maybe let’s not go there for this
analogy), or Green Mountain coffee at a different location. There is variation
in pricing and convenience. There is variation in the condiments we can use to
doctor our coffee. An economy and series of markets exist which allow Starbucks
to gather resources from dozens of other companies to provide your coffee. But,
ultimately, we are all buying coffee which has caffeine which has a desired
effect. We can go to a free AA meeting or to a soup kitchen and get some pretty
basic coffee if we don’t have the money to pay. The point is we have choices
and we can pay a high price, a low price, or no price and we get coffee.
The Starbucks model does work for a certain sector
of the population but not everyone. Since vast majority of cancer care in the
US is private, the Starbucks model falls down because we don’t actually have
any free options as a society and “low-cost healthcare” is not typically
appealing to most Americans with cancer because they have their mortality at
stake (no one wants cancer nor does anyone want to die from cancer). In fact,
desperation in the face of cancer is what makes the US one of the only places
in the developed world where people go bankrupt trying to be treated for
cancer. The ultimate inequity is that cancer care is “pay to play” in the US
and there essentially aren’t safety nets for any populations that can’t pay
(homeless) or are living below a certain income threshold (i.e., the ~10% of
Americans without healthcare plus a large percentage with insufficient
insurance).[v]
Please remember, these are human beings and they
didn’t choose to get cancer (there is no demand for cancer… there is only
demand for cancer care!). Since they didn’t have a choice in the disease they
have to be burdened with, why is there an expectation that they should pay for
the treatment? Moreover, if a patient has a stage I cancer, easily surgically
removed and cured vs. a Stage III cancer requiring months of various therapies
at a very high cost, how do we ethically explain an increased cost for a worst
state of disease? It’s really an inverse quality spectrum and we make patients
pay more for getting a lot less. We pay for insurance in case we ever do get
cancer (or other major disease). It’s a risk reduction or risk aversion
pre-payment. Like we do with our car or our house or our boat. Those last three
things we choose to have (and are luxuries). We don’t get to choose to have
health. It’s just an inherent part of being human so holding someone
accountable for it because they didn’t have the resources to “prepare for the
worst” is really the wrong attitude. Our healthcare system isn’t perfect but
there are gaps that could be easily filled if resources are allocated
efficiently to meet the whole populations needs—that’s the benefit of having a
large resource supply into the system. We just have to find the operational
efficiency to make the costs work.
However, when we remove the luxuries of insurance, Medicare, and Medicaid and other payments systems from the health sector or, worse, simply assume the government’s role is to provide healthcare 100% free to all citizens in a resource-limited or resource-constrained setting, we suddenly have an untenable situation. The economy and tax-base are not there to create the resources. We find overworked, underpaid, and undersupplied medical staff working in crowded conditions. For single entity care (e.g., HIV, tuberculosis, malaria), vertical programs have made great strides in combatting these diseases even in some of the poorest countries in the world. But cancer is anything but simple with the complexity of cross-discipline collaboration, spectrum of disease, range of treatments, and inherent costs creating huge gaps in the delivery of cancer care. Economic and physical infrastructure for the provision of care is what is needed to meet this challenge. Our current Starbucks model in the US would be extremely difficult to replicate in a low-resourced setting due to the lack of infrastructure. However, when this infrastructure is assessed, planned for, and implemented, cancer care can be delivered in these settings at a significantly lower cost per patient. Adding infrastructure implementation high-quality private facilities and public-private partnerships creates a way forward to pump resources into the system and insure that no patient is left behind. To round out this allegory, AAA locations (a commercial car-servicing company) in various parts of the US allow one to renew your driver’s license with them, rather than the DMV. I did this once, it was VERY fast, friendly, and efficient. This type of public-private partnership worked for me and I believe it will work for cancer if we are willing to try.
[ii] “Fast Facts on US Hospitals”.
Aha.org. Retrieved December 1, 2016.
[iii]
Himmelstein DU, Woolhandler S (March 2016). “The Current and Projected
Taxpayer Shares of US Health Costs”. American Journal of Public Health.
106 (3): 449–52. doi:10.2105/AJPH.2015.302997. PMC 4880216. PMID 26794173.
Government’s share of overall health spending was 64% of national health
expenditures in 2013
[iv] ^
Leonard K (January 22, 2016). “Could Universal Health Care Save U.S.
Taxpayers Money?”. U.S. News & World Report. Retrieved July 12, 2016.
-Dan Milner, MD, MSc, spent 10 years at Harvard where he taught
pathology, microbiology, and infectious disease. He began working in
Africa in 1997 as a medical student and has built an international
reputation as an expert in cerebral malaria. In his current role as
Chief Medical officer of ASCP, he leads all PEPFAR activities as well as
the Partners for Cancer Diagnosis and Treatment in Africa Initiative.
We have reviewed from start to
finish the next generation sequencing wet bench process, data review and
troubleshooting. I’d like to take a more
in-depth look at the types of variants that can be detected by the targeted
amplicon NGS panels that our lab performs:
single nucleotide variants, multi-allelic variants, multi-nucleotide
variants, insertions (including duplications), deletions and complex indels. In our lab, we review every significant
variant and variant of unknown significance in IGV to confirm the call is made
correctly in the variant caller due to the difficult nature of some of these
variants. I have included screenshots of
the IGV windows of each of these types of variants, to show what we see when we
review.
Single Nucleotide Variants (SNV)
The most common (and straight forward) type of variant is a single nucleotide variant – one base pair is changed to another, such as KRAS c.35G>A, p.G12D (shown below in reverse):
Multi-allelic Variants
A multi-allelic variant has more than one change as a single
base pair (see below – NRAS c.35G>A, p.G12D, and
c.35G>C, p.G12A – shown below in reverse). This may be the rarest type of variant – in
our lab, we have maybe seen this type in only a handful of cases over the last
four years. This could be an indication
of several clones, or different variants occurring over a period of time.
Multi-nucleotide Variants (MNV)
Multi-nucleotide variants are variants that include more
than one nucleotide at a time and are adjacent.
A common example is BRAF p.V600K (see below – in reverse) that can occur
in melanoma. Two adjacent nucleotides
are changed in the same allele. These
variants demonstrate one advantage NGS has over dideoxy (Sanger) sequencing. In dideoxy sequencing, we can see the two
base pair change, but we cannot be certain they are occurring on the same
allele. This is an important distinction
because if they occurred on the same allele, they probably occurred at the same
time, whereas, if they are on different alleles, they were probably two
separate events. It is important to know
for nomenclature as well – if they are on the same allele, it is listed as one
event, as shown below (c.1798_1799delGTinsAA, p.V600K) as opposed to two separate
mutations (c.1798G>A, p.V600M and c.1799T>A, p.V600E). As you can see in the IGV window below, both
happen on one strand.
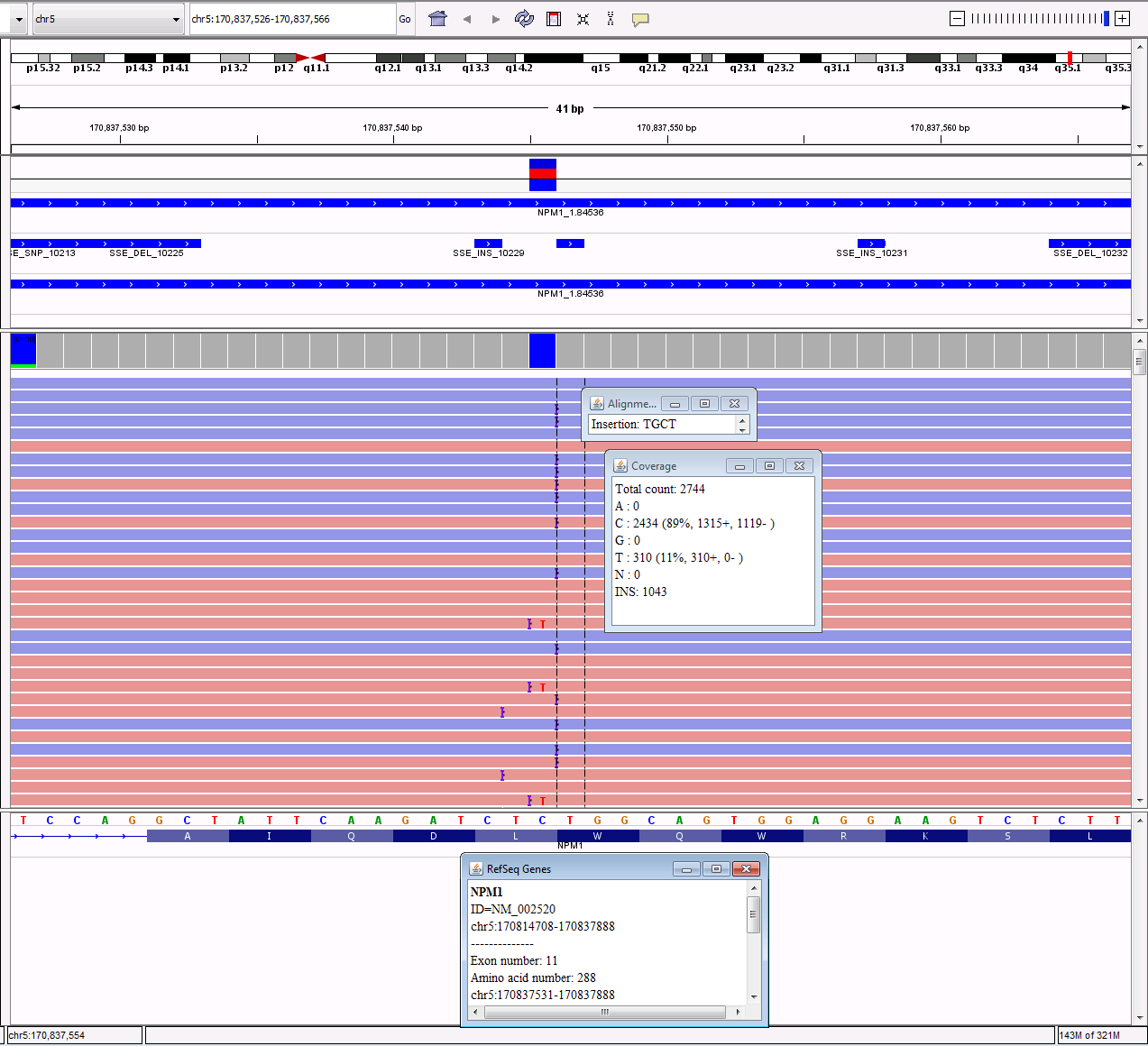
Insertions/Duplications
Insertions are an addition of nucleotides to the original
sequence. Duplications are a specific
type of insertion where a region of the gene is copied and inserted right after
the original copy. These can be in-frame
or frameshift. If they are a replicate
of three base pairs, the insertion will move the original sequence down, but
the amino acids downstream will not be affected, so the frame stays the
same. If they are not a replicate of
three base pairs, the frame will be changed, causing all of the downstream
amino acids to be changed, so it causes a frameshift. A common example of a frameshift insertion is
the 4bp insertion in NPM1 (c.863_864insCTTG, p.W288fs)
that occurs in AML. In IGV, these are
displayed by a purple hash that will show the sequence when you hover over it.
Deletions
Deletions, on the other hand, are when base pairs are
deleted from the sequence. These can be
in-frame or frameshift, as well. An
example is the 52bp deletion (c.1099_1150del, p. L367fs) found in the CALR
gene in cases of primary myelofibrosis or essential thrombocythemia.
Complex Indels
Lastly, NGS can detect complex indels. These, again, are a type of variant that we
could not distinguish for sure using dideoxy sequencing. We would be able to detect the changes, but
not whether or not they were occurring on the same strand, indicating the changes
occurred at the same time. The first
example is a deletion followed by a single nucleotide change – since these both
occur on the same strand, they most likely occurred together, so they are
called one complex deletion/insertion event (KIT c. 1253_1256delACGAinsC, p. Y418_D419delinsS). First the ACGA was deleted, then a C was
inserted.
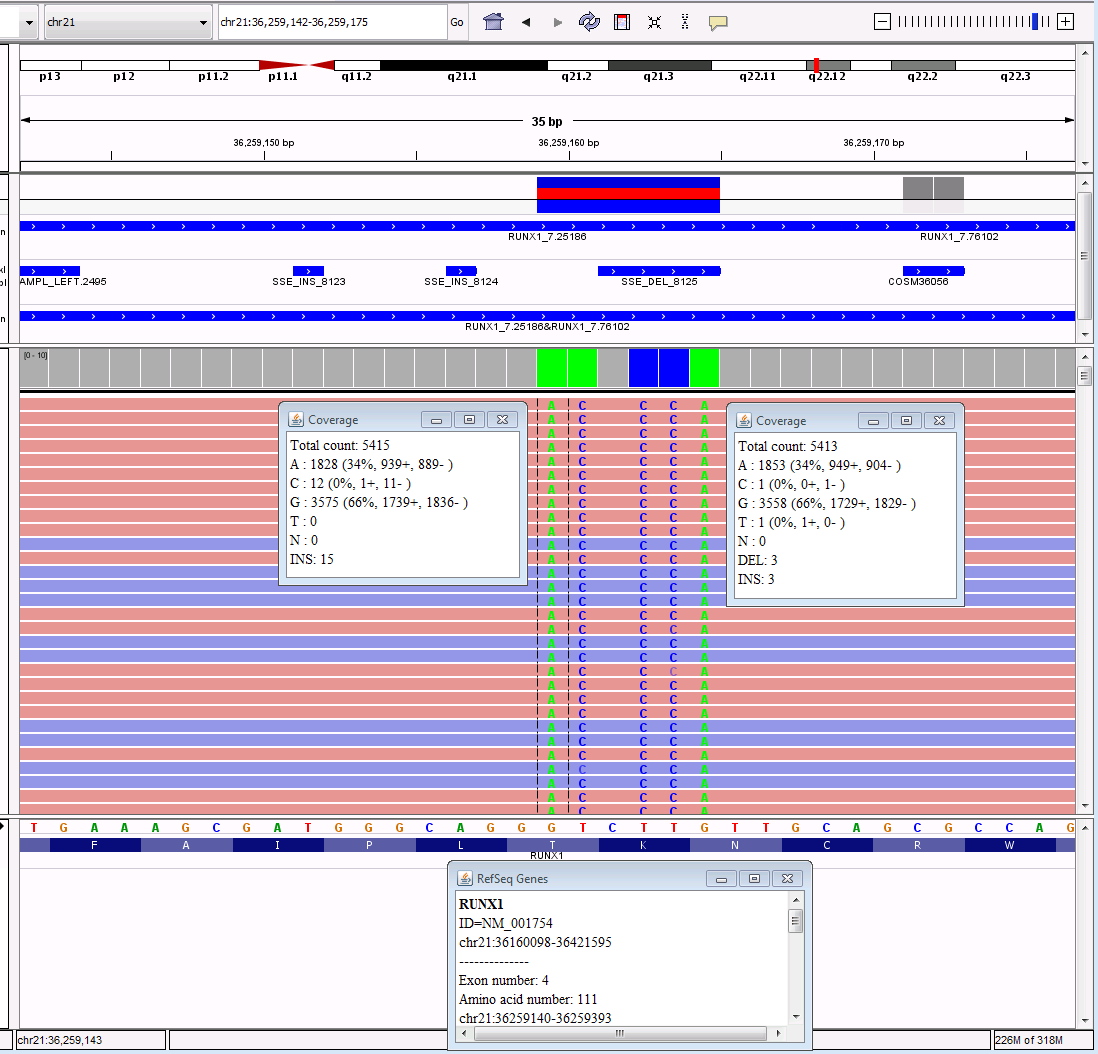
The last example involves multiple nucleotides changes all
in the same vicinity (IGV is in reverse for this specimen as well). Using HGVS nomenclature as in all the
previous examples, this would be named RUNX1 c.327_332delCAAGACinsTGGGGT,
p.K110_T111delinsGV.
-Sharleen Rapp, BS, MB (ASCP)CM is a Molecular Diagnostics Coordinator in the Molecular Diagnostics Laboratory at Nebraska Medicine.
A sixty nine year old female who underwent right breast reconstruction about 13 years ago due to breast cancer presents to the doctor office with right breast pain and right breast enlargement over the last two months. She has lost some weight and does not recall any trauma to this area. She had a textured saline implant. Examination reveals no definite palpable masses. MRI of right breast showed intact saline implant with moderate amount of fluid surrounding the implant within the intact external capsule. No adenopathy was noted. Right breast implant was removed and complete capsulectomy was performed.
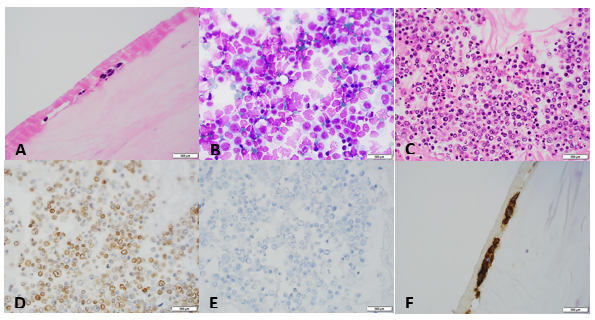
Image 1. A. Section of breast capsule with rare atypical hyperchromatic cells (arrow). B. Cytospin preparation of the fluid surrounding the implant with numerous atypical lymphocytes. C. Cell block of the fluid with large atypical lymphocytes. D, E. Lymphocytes are positive for CD30 (image D) and negative for ALK-1 (image E). F. CD30 positive cells in the section of the implant.
Diagnosis
Breast implant-associated
anaplastic large cell lymphoma.
Discussion
Breast implant associated
anaplastic large cell lymphoma is a provisional entity that is morphologically
and immunophenotypically similar to ALK-negative anaplastic large cell lymphoma.
It arises primarily in association with a breast implant. It is a very rare
entity with an incidence of 1 in 500,000 to 3 million women with implants. Tumor
cells may be localized to the seroma cavity or may involve pericapsular fibrous
tissue. Sometimes it can form a mass lesion. Locoregional lymph node may be
involved. The mean patient age is 50 years. Most patient presents with
stage 1 disease, usually with peri-implant effusion. The mean interval from
implant placement to lymphoma diagnosis is 10.9 years. There is no association
with the type of implant. Histologic examination shows two different types of
proliferations. In patients with seroma, the proliferation is confined to the
fibrous capsule (“in situ” iALCL). However, the distribution of neoplastic
lymphocytes could be heterogeneous with some cellular areas with numerous large
pleomorphic cells of varying size and some fibrotic areas with rare atypical
lymphocytes. It is beneficial to look at the seroma fluid in addition to
capsule sections, because sometimes the neoplastic lymphocytes are
predominantly present in fluid (as in our case). Patients presenting with tumor
mass show more heterogeneous proliferations infiltrating surrounding tissues
(“infiltrative” iALCL). They consists of either sheets are clusters of large
neoplastic cells accompanied by a large number of eosinophils. By immunohistochemistry,
the tumor cells are strongly positive for CD30. CD2 and CD3 are more often
positive than CD5. CD43 is almost always expressed. Most cases are CD4 positive. The
prognosis is very good in patients with disease confined to the capsule.
The median overall survival is 12 years. However, patients with a tumor mass
could have a more aggressive clinical outcome.
References
1. Swerdlow SH, Campo E, Harris NL, et al. WHO Classification of Tumours of
Haematopoetic and Lymphoid Tissues (Revised 4th edition). IARC: Lyon
2017.
2. Jaffe, E , Arber, D, et al. Hematopathology (second edition) 2017.
-Junaid Baqai, MD, was born in Chicago, IL but spent most of his life in Karachi, Pakistan. He graduated from DOW Medical College in Pakistan and did his residency in anatomic and clinical pathology at Danbury Hospital, CT followed by hematopathology fellowship from William Beaumont Hospital, Michigan and oncologic-surgical pathology fellowship from Roswell Park Cancer Institute, New York. He currently serves as Medical Director of hematology, coagulation and flow cytometry at Memorial Medical Center and Medical Director of Laboratory at Taylorville Memorial Hospital.
A 44 year old male with history of cocaine use presented with 1 year history of headache and progressive frontal lobe syndrome, including symptoms like apathy, personality changes, lack of ability to plan, poor working memory for verbal information or spatial information, Broca aphasia, disinhibition, emotional lability, etc. CT scan found extensive destruction of osteocartilaginous structures of the nasal cavity and MRI showed extensive edema of the frontal lobe. Biopsy showed chronic inflammation but negative for granulomatous inflammation. Patient’s CSF laboratory analysis was normal but ANCA was tested positive, in a P-ANCA pattern without MPO detectable. Patient was diagnosed as CIMDL. After stopping cocaine use, patient was doing better but still has mild frontal lobe syndrome.
Discussion
Anti-neutrophil cytoplasmic antibody (ANCA) are a group of
autoantibodies that directed toward antigens expressed mainly in neutrophil
granulocytes, such as proteinase 3 (RP3) and myeloperoxidase (MPO). The
presence of ANCA is mainly associated with a distinct form of small vessel
vasculitis, known as ANCA-associated vasculitis, but is also detected in other
disease, like autoimmune hepatitis, primary sclerosing cholangitis, ulcerative
colitis, and other chronic inflammatory disease. The gold standard laboratory
method to screen ANCA is indirect immunofluorescence assay (IFA or IIF), which qualitatively
capture antibodies in serum/or plasma bound to fixed human neutrophil
granulocytes.
Two form of ANCA-associated vasculitis, granulomatous with
polyangiitis (GPA) and eosinophilic granulomatous with polyangiitis (EGPA), are
systemic diseases that commonly associated with necrotizing granulomatous
vasculitis. GPA has a primary involvement of the upper and lower respiratory
tract and kidney. Autoantibodies to PR3 are found in 90% of active GPA cases,
which generates a cytoplasmic-ANCA (C-ANCA) pattern on ANCA IFA test. EGPA is a
rare form of systemic necrotizing vasculitis characterized by asthma and
eosinophilia. A perinuclear-ANCA (P-ANCA) IFA pattern directing towards MPO
antibody are often seen in EGPA cases.
Both GPA and EGPA may also present with sinonasal
involvement, causing non-infectious inflammatory lesions of the sinonasal
tract. Sinonasal inflammatory disease can also result from bacterial and fungal
infections, or other non-infectious process, such as sarcoidosis,
polychondritis, or obstruction. ANCA is detected in the majority of GPA and
EGPA case, therefore it provides useful information in differential diagnosis
of sinonasal inflammatory disease. Both GPA and EGPA are autoimmune diseases,
corticosteroids and immunosuppressive agents are effective treatment.
Sinonasal inflammation can also been seen in a subset of
patients with cocaine abuse, who normally present with midline destructive
lesions, known as cocaine-induced midline destruction lesions (CIMDL). Long-term
cocaine use has been associated with ischemia of mucosal tissue, cartilage and
bone, and cocaine abuser using intranasal inhalation route can have midline
deformity and septal perforation. Interestingly, ANCA are also found in a large
portion of CIMDL, and in contrast to GPA or EGPA, ANCA in CIMDL are primarily
directed against neutrophil elastase, generate a P-ANCA or atypical P-ANCA
pattern, without detection of MPO. Therefore, ANCA serology testing could help
the differentiation between CIMDL and GPA although these two can overlap
clinically and histopathologically. Also, CIMDL does not respond well to
immunosuppressive therapy and only consistent removal of stimuli (cocaine) can
halt the disease process.
References
Montone KT. Differential Diagnosis of Necrotizing Sinonasal Lesions. Arch Pathol Lab Med. 2015 Dec;139(12):1508-14. doi: 10.5858/arpa.2015-0165-RA.
Trimarchi M, Bussi M, Sinico RA, Meroni P, Specks U. Cocaine-induced midline destructive lesions – an autoimmune disease? Autoimmun Rev. 2013 Feb;12(4):496-500. doi: 10.1016/j.autrev.2012.08.009. Epub 2012 Aug 24.
Madani G, Beale TJ. Sinonasal inflammatory disease. Semin Ultrasound CT MR. 2009 Feb;30(1):17-24.
Timothy R. Helliwell Non-infectious Inflammatory Lesions of the Sinonasal Tract. Head Neck Pathol. 2016 Mar; 10(1): 32–39.
-Xin Yi, PhD, DABCC, FACB, is a board-certified clinical chemist,
currently serving as the Co-director of Clinical Chemistry at Houston
Methodist Hospital in Houston, TX and an Assistant Professor of Clinical
Pathology and Laboratory Medicine at Weill Cornell Medical College.
A 24 year old male with no past medical history presented with
fevers, myalgia, and cough following return from a 1-week trip to Guatemala
where he spent significant time within caves. The patient described his cough
as persistent, non-productive, and associated with mild shortness of breath at
rest that significantly worsens with activity. In the emergency department, the
patient was afebrile with a WBC of 10.2, Transaminitis, and chest X-ray showed
diffuse reticular pattern. He underwent a bronchoscopy and BAL washout.
Laboratory Findings
Histoplasmosis Urine Antigen test came back positive.
Image 1. Fungal culture with white/tan, fluffy mold (growth at day 7).Image 2. Scotch tape prep with tuberculate macroconidium. This mold was morphologically identified as Histoplasma capsulatum and sent to Mayo Laboratories for further confirmatory testing.
Discussion
Histoplasma
capsulatum is an intracellular, thermally dimorphic fungus (grows as a
yeast at body temperature/37°C in humans or culture media and as mold at 25°C
in the environment/culture media). Histoplasma
is found in soil, particularly in areas containing bird and bat droppings,
such as caves. Within the United States Histoplasma
in found in central and eastern states with a predominance in the Ohio and
Mississippi River Valleys. This fungus is also found in parts of Central and
South America, Africa, Asia, and Australia.
Infection with
Histoplasma capsulatum causes
significant morbidity and mortality worldwide. Upon inhalation of conidia, H. capsulatum transforms into the
pathogenic yeast phase. This form replicates within macrophages that carry the
yeast from the lungs to other organs. Histoplasmosis has three main forms:
Acute
primary histoplasmosis which presents as a pneumonia with fever, cough,
myalgia.
Chronic
cavitary histoplasmosis which is characterized by pulmonary lesions that often
resemble cavitary tuberculosis.
Progressive
disseminated histoplamosis that spreads to infect many organs in
immunocomprimised patients.
In the laboratory, culture of blood, tissue and respiratory
specimens may be completed. In addition, a test for H. capsulatum
antigen is sensitive and specific when simultaneous serum and urine specimens
are tested. It is important to note that cross-reactivity with other fungi (Coccidioides
immitis, Blastomyces dermatitidis, Paracoccidioides brasiliensis,
Penicillium marneffei) has been identified.
Growth on fungal culture shows white/tan, fluffy mold
that turns to brown to buff with age. The organism may also produce wrinkled,
moist, or heaped yeast-like colonies that are soft and cream when grown at 37°C
on certain media. Scotch tape preparation of the mold form shows tuberculate
macroconidia, a diagnostic structure of Histoplasma
capsulatum. The mycelia are septate and produce microconidia and
macroconidia. Yeast forms of Histoplasma
capsulatum are small (2 to 4 μm) and reproduce by budding. These budding
forms may be seen on histology specimens. A commercially available DNA probe
can be performed on culture material to confirm identification.
Patients with mild-moderate histoplasmosis can often have
resolution of their symptoms without treatment. Those with more moderate-severe
disease require antifungal agents including amphotericin B or itraconazole.
-Nicole Mendelson, MD
is a 1st year Anatomic and Clinical Pathology resident at the
University of Vermont Medical Center.
-Christi Wojewoda, MD, is the Director of Clinical Microbiology at the University of Vermont Medical Center and an Associate Professor at the University of Vermont.
Nichole Baker, PA, works at Mercy Regional Medical Center in
Durango, Colorado as a Pathologists Assistant. Nichole started working as a
volunteer with Mbarara University of Science Technology (MUST) in 2017 through
the existing partnership with Massachusetts General Hospital Pathology
department– she responded to an advertisement looking for pathology volunteers
that Dr.
Drucilla Roberts had placed in a pathology journal. She decided to visit
the laboratory and see in what ways she could help. In total, she has visited
three times, and in that time, has accomplished incredible things! What was
particularly impressive to me is that Nichole single-handedly solved a very
complex problem in the MUST laboratory. In fact, it was this same problem that
many people (including myself!), had attempted to solve and could not find the
means to do so! Not only did she implement a solution to the problem, but she
did it in just two weeks!
Read on to hear from Nichole about her experience making
positive changes in her global community. I guarantee you will be inspired by
her work, her enthusiasm, and her can-do attitude!
Q: Nichole, I know that you recently returned from
Uganda, and you were able to team up with the pathology staff at MUST to make
some major changes and implemented a solution to a major problem. Can you tell
me about your project?
A: It started with realizing from my two prior visits
to the anatomic pathology lab at MUST that the laboratory had a faulty internal
tracking system for cases. This had two consequences: The first is that the
case turn-around time has been very difficult to track. This even results in
occasional cases being lost entirely. The second is that there is no repository
of cases to be able to easily conduct research.
What I decided to do was build a free computer program that
could accession the cases, track them, generate a pathology report, and give a
report of turn-around-time. Not having a computer science background myself, I
contacted a friend who connected me with a software engineer in Denver,
Colorado. He helped guide me in what would be feasible to accomplish and helped
me find a pro-bono programmer based in Belize named Maurice, who had some
background in healthcare IT. We started building the system less than a month
away from my departure to Uganda.
My goal was to work with the laboratory staff to build a
program based around their needs, for which I needed to be there in person to
clearly identify – I set out on my third trip in March 2019, this time for two
weeks. Maurice and I built a cloud-based tracking program and every day, we
would try it out in the laboratory. Day by day, issues would arise, such as the
need to add a sign-out function, general localization changes, or adding a
timestamp for a particular function. Fortunately for me, Maurice and I had a
substantial time difference which really worked to our advantage. I would try
the system out during the day and then email Maurice a report that he would
just be waking up to. He was able to work in Belize while we were asleep in
Uganda, and when I returned to the lab the next day, the program had been
updated with the changes. This allowed for rapid progress and the pathology
staff grew more and more excited to use the system as it improved. So, day by
day, we made the program better and better.
Q: What were some of the unique challenges that you faced
when implementing the program?
A: Originally, we had planned to use a laptop with
boosted RAM to act as a local server, but the network in the hospital wasn’t
functioning as needed. On-site we realized we’d need to shift to an internet
server and to do so we had to improve the internet access in the laboratory in
order to run the program –this was difficult because IT progress can be slow
in Uganda.
Another example that is unique to this setting is the difficulty we had in generating unique patient identifiers in the registration system. In the US, two patient identifiers are required for each sample, and that is easy to obtain because everyone knows their date of birth. In Uganda, things are not as clear and straightforward. We might only have the village they live in, or a phone number. We had to look to see what items were most consistently reported and use those.
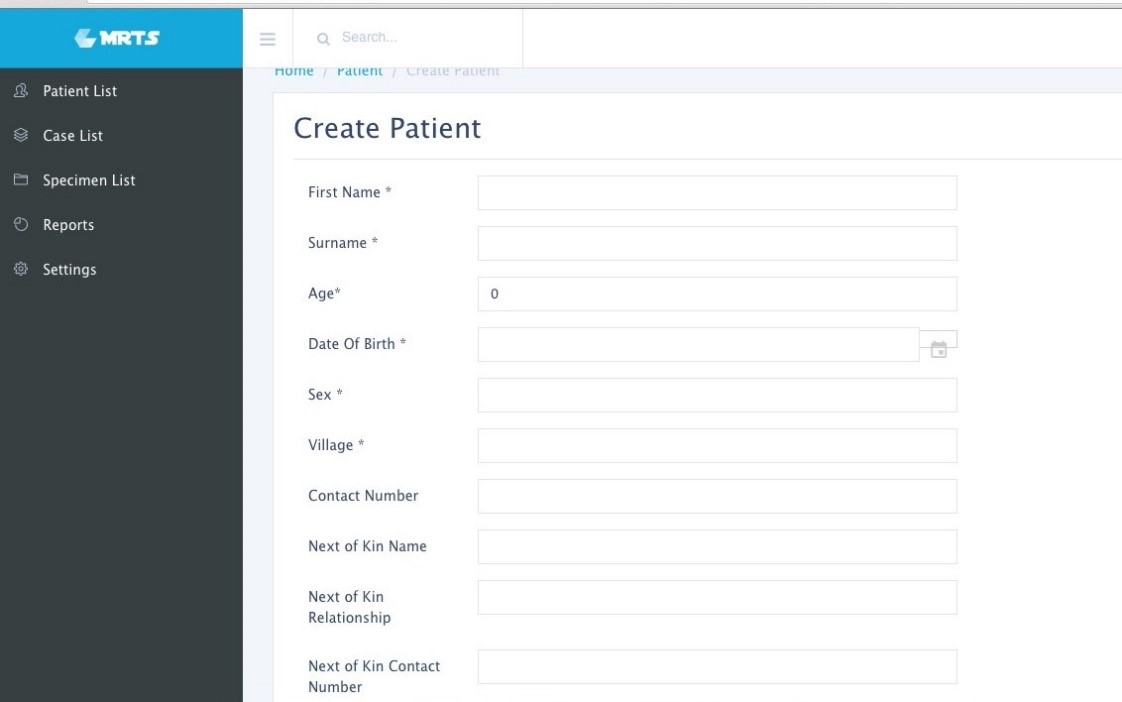
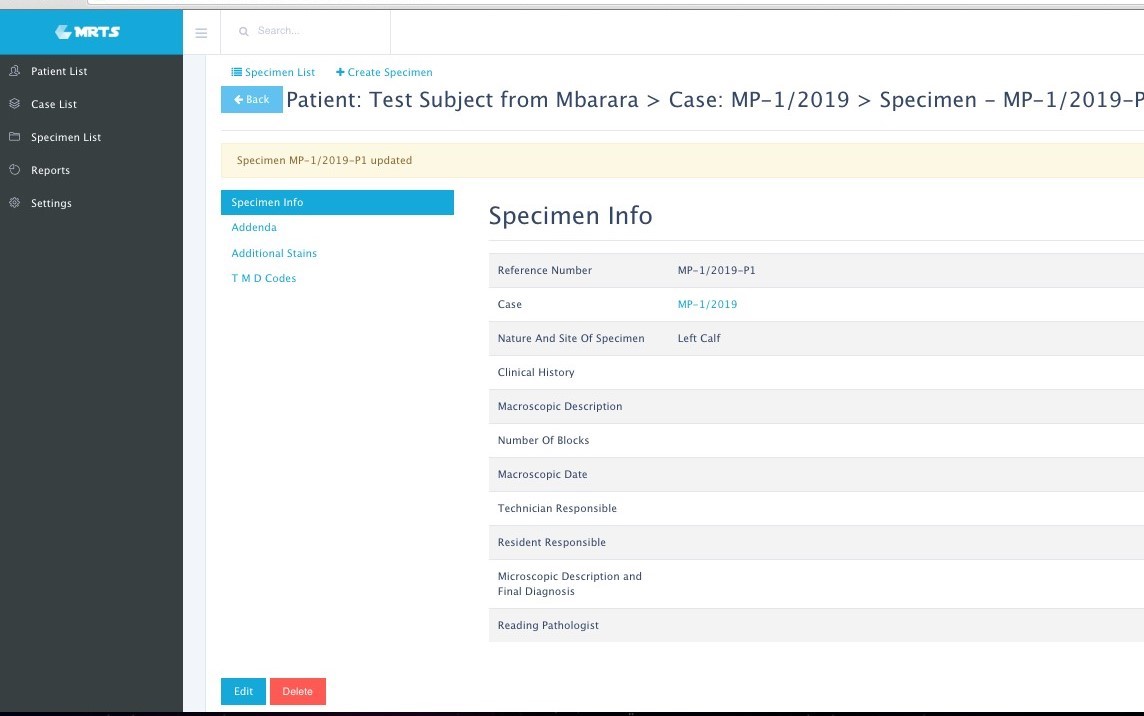
Image 1. Sample patient intake page. Patients can be uniquely identified and stored for future visits. Image 2. Sample case information page. All aspects of the case can be stored and easily retrieved, including IHC performed, and diagnosis codes.
Q:How are you financing the data storage and
internet?
A: All fees and
costs associated with the program were raised by a small charity organization I
started in 2017 called “Path of Logic”
which has 501c3 status, making any donations tax deductible. With the funds raised,
a shared laboratory laptop was purchased. We are using a cloud-based system
that charges based on storage space. Right now, the storage need is low because
reports are stored as PDFs, but we may need to expand in the future. The
internet connection is also a low expense, as it’s simply a backup modem that’s
used when the university internet is not functioning.
Q: It’s now been two months since you rolled out the
program in the lab. What results have you seen from that?
A: Once we got going, we have been able to identify
where the delays were in processing the cases. After I returned back to the US,
Maurice and I continued to work on small issues remotely, such as single vs.
double click preferences and those sorts of things.
So far, 421 cases have been
registered in the system. The average turnaround time is 12.5 days. We still have a lot of work to go, but this is
the first we’ve been able to track this number. Many of the cases that were
started in the weeks following my departure were not signed out, but as the
team sees the value in the system, the more accurate that average will be,
allowing adjustments to be made accordingly.
We also added in the ability to assign ICD codes to the
final diagnosis to allow for a way to categorize the cases to make the
diagnosis searchable. Now we are going to be able to generate epidemiological
data. This feature is not yet in use by the pathology team, but we are hopeful
that as the system becomes more routine, this will be the next step to incorporate.
Image 3. Sample Final Pathology Report, stored as a PDF.
Q: What future impact do you think this program might
have?
A: In addition to being able to easily track cases, build pathology reports, generate icd codes for researching cases more easily, we also hope that this will eventually result in increased funding for pathology services in Uganda. Right now, the money allocated from the Ugandan Ministry of Health is going towards HIV, malaria, and cancer treatment – but not for diagnostics. The Department of Education allows some funds for Pathology, but only about 30% of what is needed. Part of the reason why is that until now, there has been no way to quantify the number of cancer cases. With our program, we will be able to generate that data to show real numbers when lobbying for increased funds.
-Dana Razzano, MD is a Chief Resident in her third year in
anatomic and clinical pathology at New York Medical College at
Westchester Medical Center and will be starting her fellowship in
Cytopathology at Yale University in 2020. She was a top 5 honoree in
ASCP’s Forty Under 40 2018 and was named to The Pathologist’s Power List
of 2018. Follow Dr. Razzano on twitter @Dr_DR_Cells.
76 year old man with a history of chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) with new anterior mediastinal mass and increasing lymphadenopathy.
Lymph Node Biopsy
H&E
Diagnosis
Tissue sections show a diffuse atypical lymphoid infiltrate
that completely effaces the normal nodal architecture. The infiltrate is
composed of numerous small lymphocytes with round to mildly irregular nuclei,
clumped chromatin, inconspicuous nucleoli and scant cytoplasm. There are also
expanded pale areas that contain intermediate sized cells with more open
chromatin and distinct single to multiple nucleoli. These cells are most
consistent with prolymphocytes/paraimmunoblasts and form the proliferation
centers characteristic of CLL/SLL. Occasional centroblastic-type B-cells are
noted within these proliferation centers. In addition, there are scattered single
to multinucleated cells that have irregular nuclear membranes with pale,
vesicular chromatin and prominent inclusion-like, eosinophilic nucleoli. These
cells morphologically resemble Hodgkin cells, Reed-Sternberg cells, mummified
forms and other variants. These large cells are more evident in areas with a
histiocyte rich background and around foci of necrosis. Occasionally, apoptotic
bodies and mitotic figures are seen.
Immunohistochemical studies
show that the vast majority of the small-intermediate lymphocytes express B-cell
markers CD20 (dim) and PAX5 and co-express CD5 and CD23 (subset). This is
consistent with a background of CLL/SLL. The large atypical cells are positive
for CD30, PAX5 and CD20 (variable). CD3 highlights numerous scattered
background small T-cells, which are increased in the areas with the large cells.
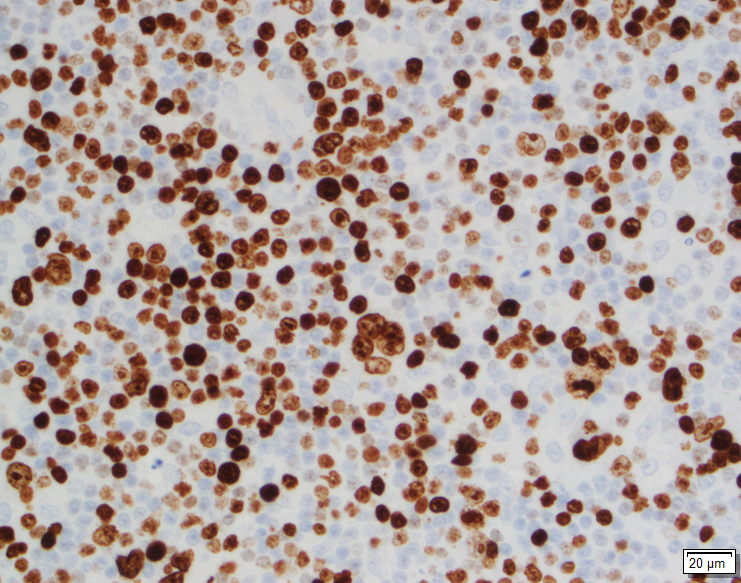
In situ hybridization for Epstein Barr viral RNA (EBER ISH) is mainly staining
the large atypical cells. By Ki-67, the proliferation fraction is overall
increased (40%) with increased uptake by the large atypical cells.
The morphologic and immunophenotypic findings are consistent
with involvement by the patient’s known small lymphocytic lymphoma/chronic
lymphocytic leukemia (SLL/CLL) with aggressive morphological features. The
aggressive features include expanded proliferation centers and an elevated
Ki-67 proliferative index (40%). Additionally there are histiocyte/T-cell rich
areas composed of multiple EBV positive large atypical cells with morphologic
and immunophenotypic features compatible with Hodgkin/ Reed-Sternberg cells.
These areas are most in keeping with evolving classic Hodgkin lymphoma. Sheets
of large cells indicative of large cell transformation are not seen, although
increased scattered large centroblastic-type B cells are present.
Discussion
Lymph node involvement by CLL/SLL will typically show a
diffuse proliferation of small lymphocytes with effacement of the normal nodal
architecture. The small lymphocytes have
round nuclei, clumped chromatin and scant cytoplasm. Scattered paler areas
known as proliferation centers are characteristic of this entity. The
proliferation centers are composed of a mixture of cell types including small
lymphocytes, prolymphocytes and paraimmunoblasts. Prolymphocytes are small to
medium in size with relatively clumped chromatin, whereas paraimmunoblasts are
larger cells with round to oval nuclei, dispersed chromatin, eosinophilic
nucleoli and slightly basophilic cytoplasm. Some cases show increased and
enlarged proliferation centers with a higher proliferation rate. This must be
distinguished from large cell transformation.1
Aggressive features of CLL/SLL include proliferation centers
that are broader than a 20x field or becoming confluent. An increased Ki-67
proliferation >40% or >2.4 mitoses in the proliferation centers can also
portend a more aggressive course. These cases tend to have worse outcomes than
typical CLL/SLL and better outcomes than cases that have undergone Richter
transformation to diffuse large B-cell lymphoma (DLBCL). Transformation to
DLBCL occurs in 2-8% of patients with CLL/SLL. Less than 1% of patients with
CLL/SLL develop classic Hodgkin lymphoma (CHL). In order to diagnose CHL in the
setting of CLL/SLL, classic Reed-Sternberg cells need to be found in a
background appropriate for CHL, which includes a mixed inflammatory background.
The majority of these CHL cases will be positive for EBV.1
Richter’s transformation is defined as an aggressive
evolution of CLL. While the most common type of transformation is to a
high-grade B-cell Non-Hodgkin lymphoma, other histological transformations have
been described. This includes CHL, lymphoblastic lymphoma, hairy cell leukemia
and high-grade T-cell lymphomas. The prognosis for patients who present with
transformation to CHL is poor compared to de novo CHL.2
A large study from the M.D. Anderson Cancer Center described 4121 patients with
CLL/SLL and found that only 18 patients or 0.4% developed CHL. The median time
from CLL to CHL diagnosis was 4.6 years. Fourteen of the patients received
chemotherapy. The overall response rate was 44% with a complete response rate
of 19%. The median overall survival was 0.8 years and all patients eventually
died from disease recurrence or progressive disease.3 This dismal
prognosis is similar to patients with Richter transformation to DLBCL and much
worse than patients with de novo CHL, which is curable in >85% of cases.1
References
Swerdlow SH, Campo E, Harris NL, et al. WHO
Classification of Tumours of Haematopoetic and Lymphoid Tissues (Revised 4th
edition). IARC: Lyon 2017.
Janjetovic S, Bernd HW, Bokemeyer C, Fiedler W. Hodgkin’s
lymphoma as a rare variant of Richter’s transformation in chronic lymphocytic
leukemia: A case report and review of the literature. Mol Clin Oncol.
2016;4(3):390–392.doi:10.3892/mco.2016.727.
Tsimberidou, AM, O’Brien, S and Kantarjian, HM,
et. al. Hodgkin transformation of chronic lymphocytic leukemia. Cancer. 2006;107(6).doi.org/10.1002/cncr.22121.
–Chelsea Marcus, MD is a Hematopathology Fellow at Beth Israel
Deaconess Medical Center in Boston, MA. She has a particular interest in
High-grade B-Cell lymphomas and the genetic alterations of these
lymphomas.