Case History
76 year old man with a history of chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) with new anterior mediastinal mass and increasing lymphadenopathy.
Lymph Node Biopsy

H&E

Diagnosis
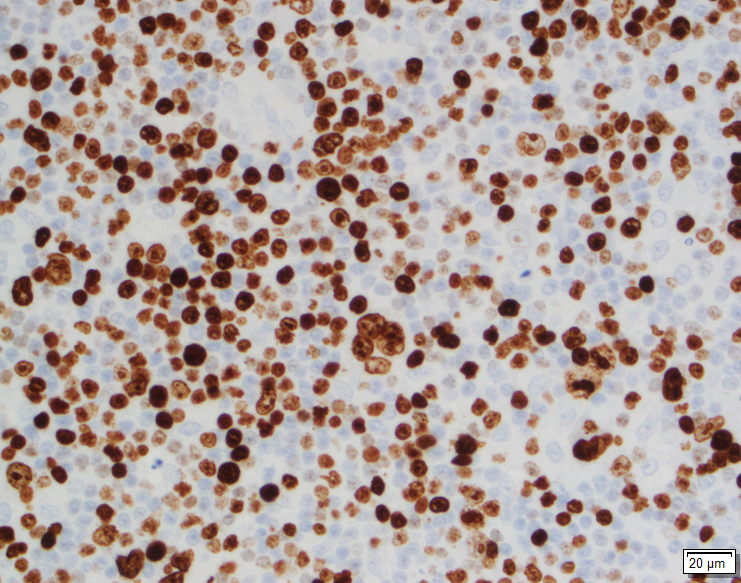
Tissue sections show a diffuse atypical lymphoid infiltrate that completely effaces the normal nodal architecture. The infiltrate is composed of numerous small lymphocytes with round to mildly irregular nuclei, clumped chromatin, inconspicuous nucleoli and scant cytoplasm. There are also expanded pale areas that contain intermediate sized cells with more open chromatin and distinct single to multiple nucleoli. These cells are most consistent with prolymphocytes/paraimmunoblasts and form the proliferation centers characteristic of CLL/SLL. Occasional centroblastic-type B-cells are noted within these proliferation centers. In addition, there are scattered single to multinucleated cells that have irregular nuclear membranes with pale, vesicular chromatin and prominent inclusion-like, eosinophilic nucleoli. These cells morphologically resemble Hodgkin cells, Reed-Sternberg cells, mummified forms and other variants. These large cells are more evident in areas with a histiocyte rich background and around foci of necrosis. Occasionally, apoptotic bodies and mitotic figures are seen.
Immunohistochemical studies show that the vast majority of the small-intermediate lymphocytes express B-cell markers CD20 (dim) and PAX5 and co-express CD5 and CD23 (subset). This is consistent with a background of CLL/SLL. The large atypical cells are positive for CD30, PAX5 and CD20 (variable). CD3 highlights numerous scattered background small T-cells, which are increased in the areas with the large cells. In situ hybridization for Epstein Barr viral RNA (EBER ISH) is mainly staining the large atypical cells. By Ki-67, the proliferation fraction is overall increased (40%) with increased uptake by the large atypical cells.
The morphologic and immunophenotypic findings are consistent with involvement by the patient’s known small lymphocytic lymphoma/chronic lymphocytic leukemia (SLL/CLL) with aggressive morphological features. The aggressive features include expanded proliferation centers and an elevated Ki-67 proliferative index (40%). Additionally there are histiocyte/T-cell rich areas composed of multiple EBV positive large atypical cells with morphologic and immunophenotypic features compatible with Hodgkin/ Reed-Sternberg cells. These areas are most in keeping with evolving classic Hodgkin lymphoma. Sheets of large cells indicative of large cell transformation are not seen, although increased scattered large centroblastic-type B cells are present.
Discussion
Lymph node involvement by CLL/SLL will typically show a diffuse proliferation of small lymphocytes with effacement of the normal nodal architecture. The small lymphocytes have round nuclei, clumped chromatin and scant cytoplasm. Scattered paler areas known as proliferation centers are characteristic of this entity. The proliferation centers are composed of a mixture of cell types including small lymphocytes, prolymphocytes and paraimmunoblasts. Prolymphocytes are small to medium in size with relatively clumped chromatin, whereas paraimmunoblasts are larger cells with round to oval nuclei, dispersed chromatin, eosinophilic nucleoli and slightly basophilic cytoplasm. Some cases show increased and enlarged proliferation centers with a higher proliferation rate. This must be distinguished from large cell transformation.1
Aggressive features of CLL/SLL include proliferation centers that are broader than a 20x field or becoming confluent. An increased Ki-67 proliferation >40% or >2.4 mitoses in the proliferation centers can also portend a more aggressive course. These cases tend to have worse outcomes than typical CLL/SLL and better outcomes than cases that have undergone Richter transformation to diffuse large B-cell lymphoma (DLBCL). Transformation to DLBCL occurs in 2-8% of patients with CLL/SLL. Less than 1% of patients with CLL/SLL develop classic Hodgkin lymphoma (CHL). In order to diagnose CHL in the setting of CLL/SLL, classic Reed-Sternberg cells need to be found in a background appropriate for CHL, which includes a mixed inflammatory background. The majority of these CHL cases will be positive for EBV.1
Richter’s transformation is defined as an aggressive evolution of CLL. While the most common type of transformation is to a high-grade B-cell Non-Hodgkin lymphoma, other histological transformations have been described. This includes CHL, lymphoblastic lymphoma, hairy cell leukemia and high-grade T-cell lymphomas. The prognosis for patients who present with transformation to CHL is poor compared to de novo CHL.2 A large study from the M.D. Anderson Cancer Center described 4121 patients with CLL/SLL and found that only 18 patients or 0.4% developed CHL. The median time from CLL to CHL diagnosis was 4.6 years. Fourteen of the patients received chemotherapy. The overall response rate was 44% with a complete response rate of 19%. The median overall survival was 0.8 years and all patients eventually died from disease recurrence or progressive disease.3 This dismal prognosis is similar to patients with Richter transformation to DLBCL and much worse than patients with de novo CHL, which is curable in >85% of cases.1
References
- Swerdlow SH, Campo E, Harris NL, et al. WHO Classification of Tumours of Haematopoetic and Lymphoid Tissues (Revised 4th edition). IARC: Lyon 2017.
- Janjetovic S, Bernd HW, Bokemeyer C, Fiedler W. Hodgkin’s lymphoma as a rare variant of Richter’s transformation in chronic lymphocytic leukemia: A case report and review of the literature. Mol Clin Oncol. 2016;4(3):390–392.doi:10.3892/mco.2016.727.
- Tsimberidou, AM, O’Brien, S and Kantarjian, HM, et. al. Hodgkin transformation of chronic lymphocytic leukemia. Cancer. 2006;107(6).doi.org/10.1002/cncr.22121.

–Chelsea Marcus, MD is a Hematopathology Fellow at Beth Israel Deaconess Medical Center in Boston, MA. She has a particular interest in High-grade B-Cell lymphomas and the genetic alterations of these lymphomas.
