Case history
A 45 year-old man presented with vomiting and diarrhea for 5 days. Laboratory studies demonstrated anemia and thrombocytopenia, an elevated ferritin level (23,772 ug/L) and methemoglobinemia. Chest roentgenography revealed cardiomegaly. A follow-up ECHO showed a desreased ejection fraction of 15%. Work-up confirmed viral myocarditis and G6PD deficiency as the cause of the cardiac findings and methemoglobinemia respectively. His clinical condition deteriorated despite therapy: he developed acute kidney and liver failure and had worsening cytopenias. A bone marrow biopsy was performed.
Histomorphological findings





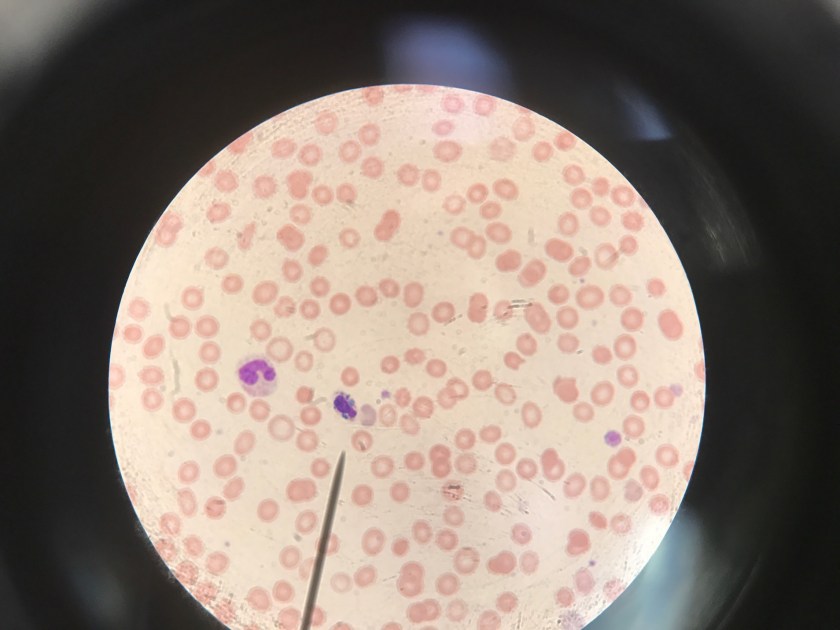
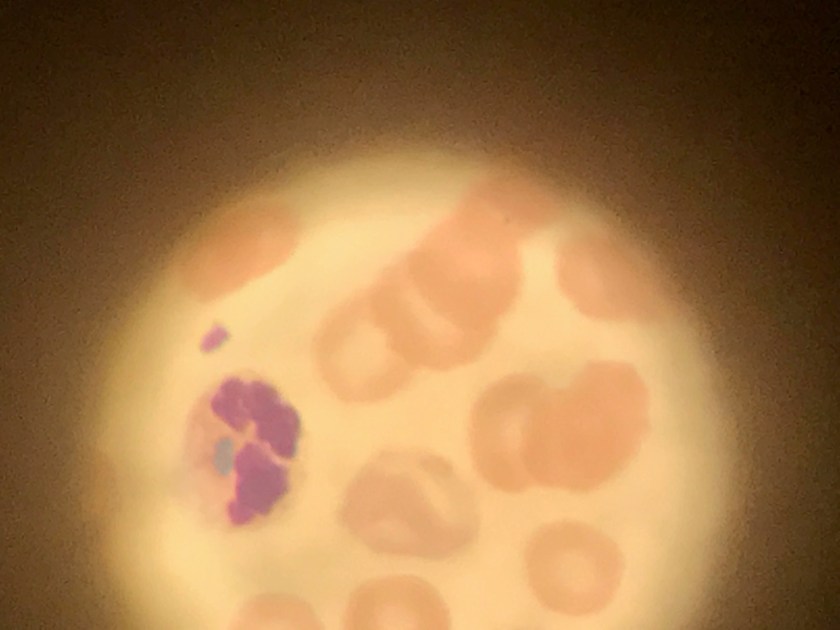
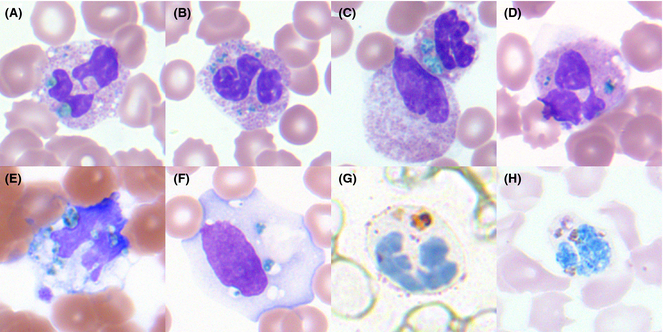
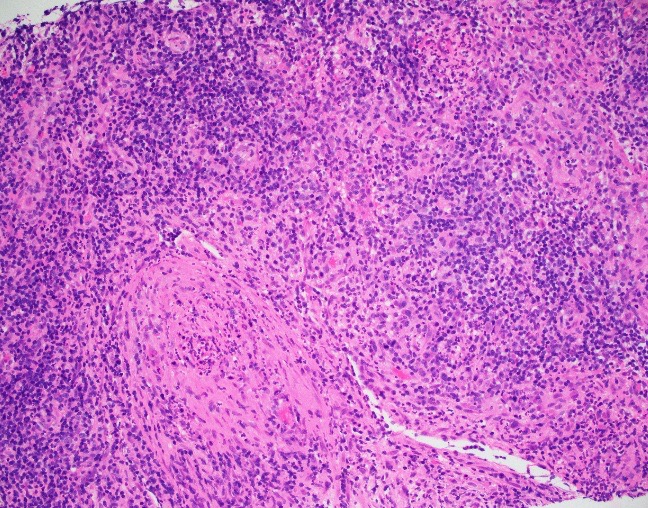
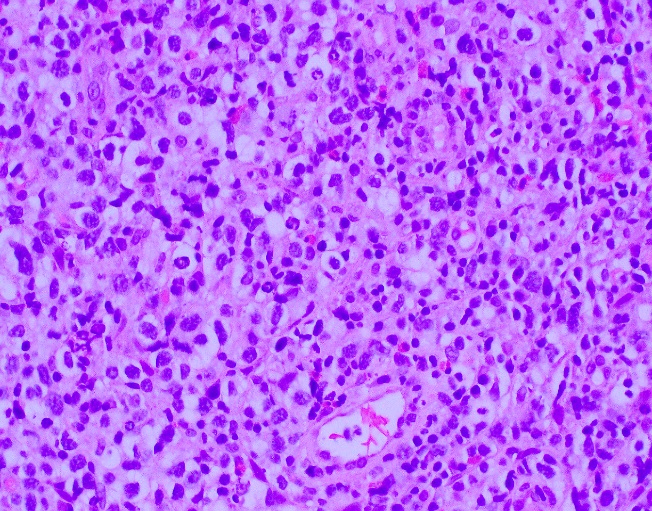
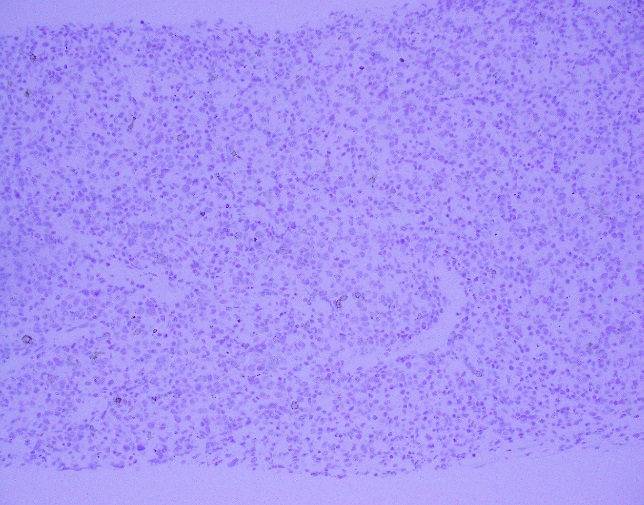
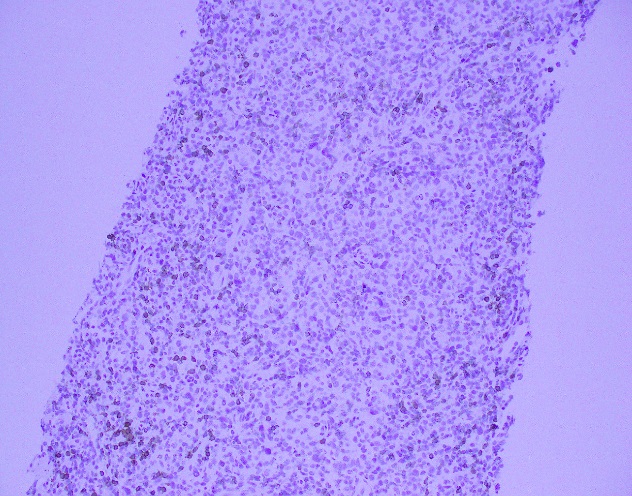
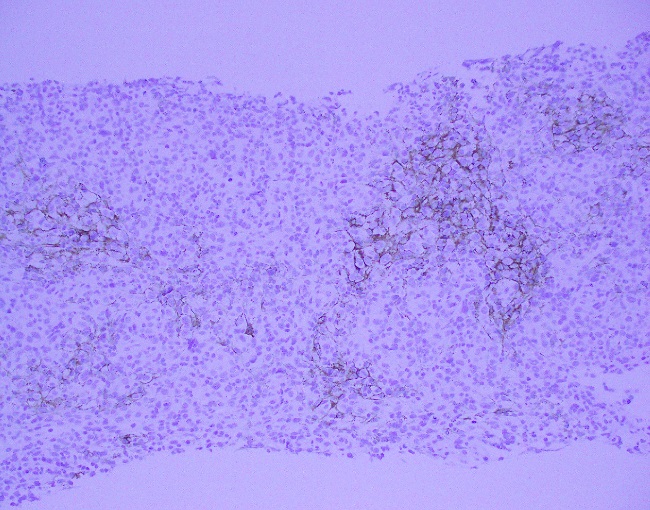
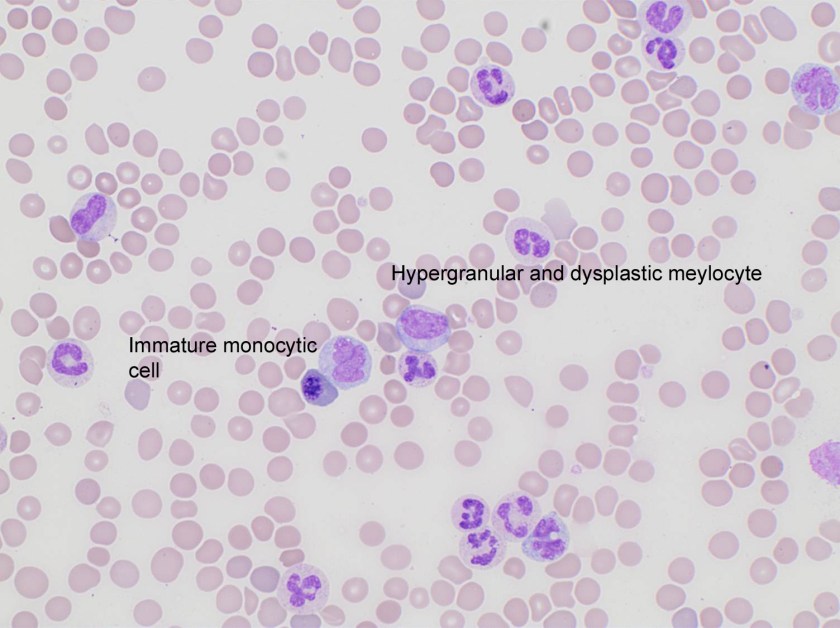
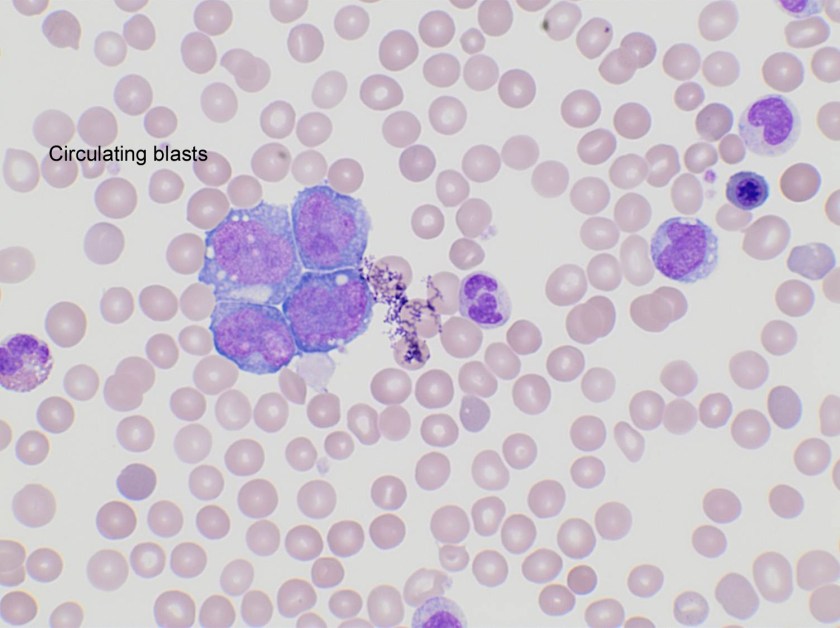
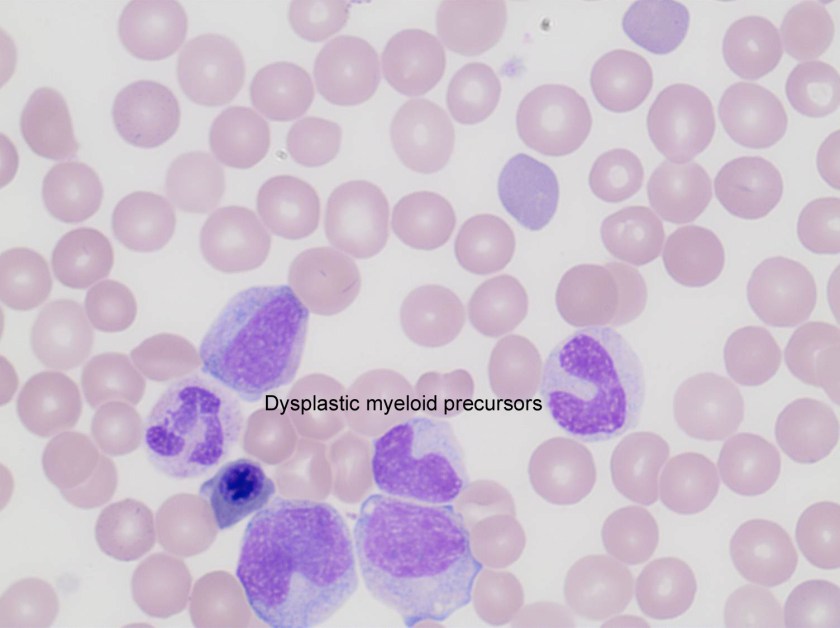
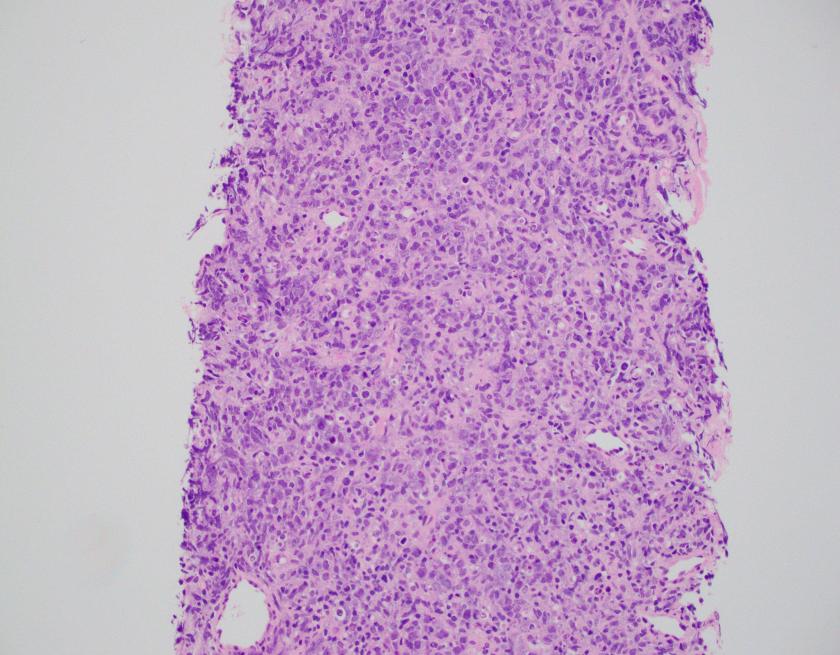
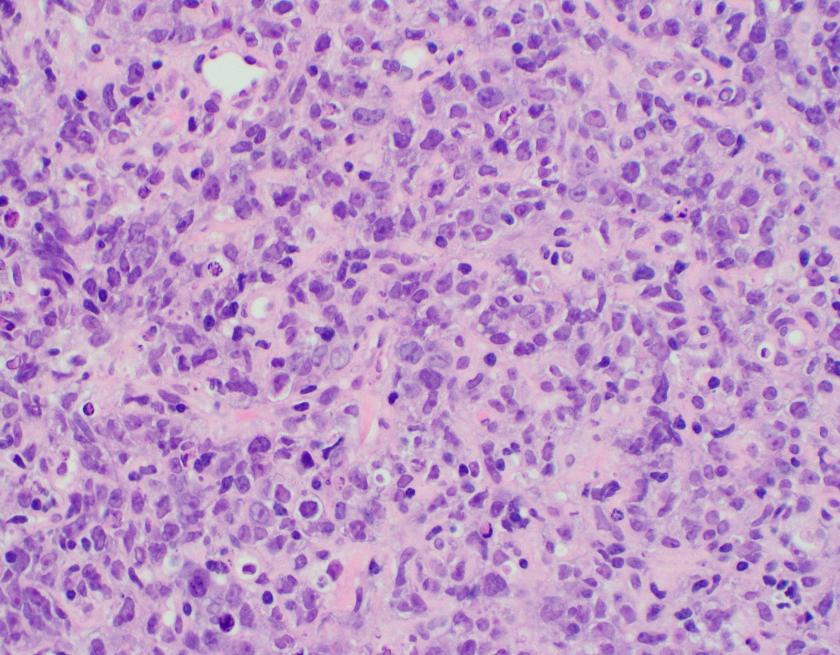
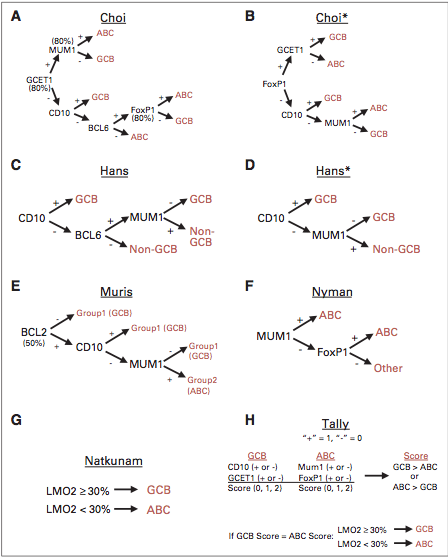
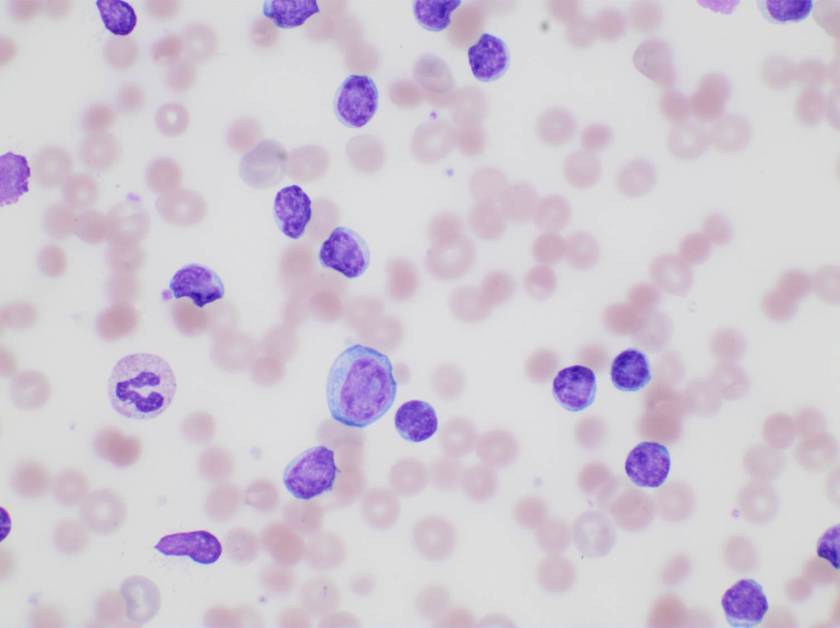
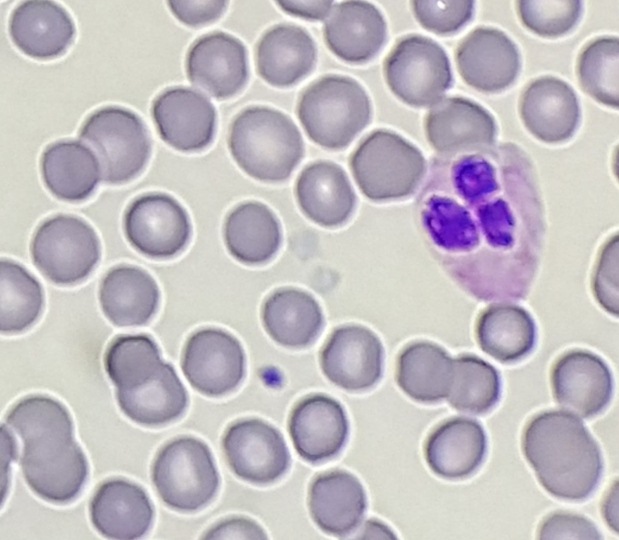
Evaluation of the peripheral blood (not pictured) confirmed a macrocytic anemia with marked anisopoikilocytosis including schistocytes, polychromasia, nucleated red blood cells, absolute neutrophilia, monocytosis and thrombocytopenia. The marrow aspirate smear demonstrated appropriate maturation in all cell lines. Scattered hemophagocytic histiocytes (pictured above) were noted. The bone core biopsy was high-normocellular for age with progressive trilineage hematopoeisis. Scattered histiocytes with internalized erythroid cells and debris were visualized. There was no increase in blasts. Flow cytometry analysis performed on the bone marrow aspirate did not show a significant increase in blast population. Gating on the lymphocytes did not show a B-cell monoclonal population or T-cell abnormality by markers assayed.
Diagnosis
High-normocellular marrow with progressive trilineage hematopoeisis and prominent hemophagocytic histiocytes.
Overall the patient met the clinical criteria (see below) for Hemophagocytic Lymphohistiocytosis (HLH); with fever (≥38.5 C), splenomegaly, bicytopenia, presence of hemophagocytic histiocytes in bone marrow and high ferritin level (>500ng/mL).
A clinical diagnosis of HLH was rendered.
Discussion
HLH is an uncommon hematologic disorder that is often fatal. The underlying pathogenesis involves an exaggerated but ineffective inflammatory response of excessive macrophage and T-cell activation, and impairment of natural killer (NK) and cytotoxic T-cell function. HLH has familial and acquired forms. Secondary, or acquired HLH can be associated with infections (especially viral etiologies), underlying malignancy (particularly lymphomas and leukemias), and medications used for systemic lupus erythematosus. Clinically, autoimmune disease-associated HLH overlaps significantly with macrophage activation syndrome (MAS).
HLH is a clinical diagnosis that can be established with molecular testing or by meeting five of eight clinical and laboratory diagnostic criteria according to the HLH-2004 guidelines.
HLH-2004: Revised diagnostic guidelines for HLH10
The diagnosis HLH can be established if one of the two criteria below is met:
- A molecular diagnosis consistent with HLH (i.e., reported mutations found in either PRF1 or MUNC13-4); or
- Diagnostic criteria for HLH are fulfilled (i.e., at least five of the eight criteria listed below are present:
- Persistent fever
- Splenomegaly
- Cytopenias (affecting ≥2 of 3 lineages in the peripheral blood):
- Hemoglobin <90g/L (in infants <4 weeks: <100g/L)
- Platelets <100 x 109/L
- Neutrophils <1.0 x 109/L
- Hypertriglyceremia and/or hypofibrinogenemia:
- Fasting triglycerides ≥3.0 mmol/L (i.e., ≥ 265mg/dl)
- Fibrinogen ≤1.5 g/L
- Hemophagocytosis in bone marrow* or spleen or lymph nodes, no evidence of malignancy
- Serum ferritin ≥ 500µg/L (i.e., 500 ng/ml)
- Low or absent NK cell activity (according to local laboratory reference)
- Increased serum sIL2Rα (according to local laboratory reference)
*In hematopathology, the finding of relevance is the presence of hemophagocytic histiocytes in the marrow or other biopsies organs. While debris-laden histiocytes are commonly noted in marrow aspirates, the findings of engulfed erythroid cells is warranted to call a ‘hemophagocytic’ histiocyte.
Often a bone marrow biopsy will be performed in cases where there is clinical suspicion for HLH. This serves to try and visualize the hemophagocytic activity, and to rule out other diseases with similar clinical presentations as HLH. The pathologic evaluation of HLH is tricky, since there is no established criteria for quantitation of hemophagocytic histiocytes in a bone marrow aspirate. Furthermore, hemophagocytosis is not specific to HLH and can be seen in other conditions such as: post-blood transfusion, chemotherapy, sepsis and major operations. Published data shows that the presence of hemophagocytosis has a sensitivity of 83% and a specificity of only 60% in diagnosing HLH.
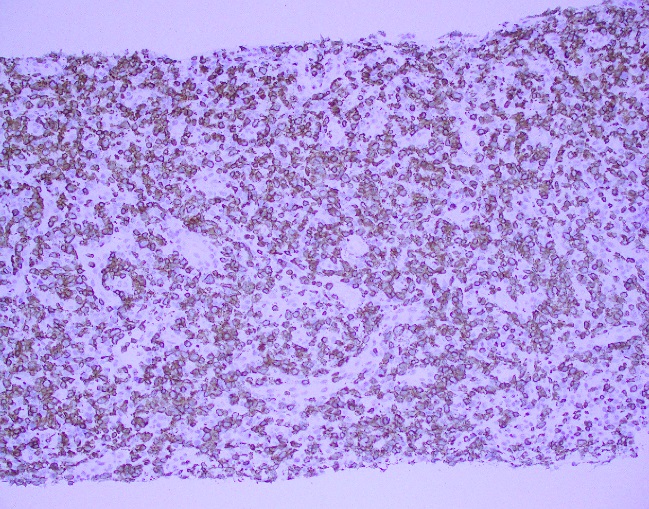
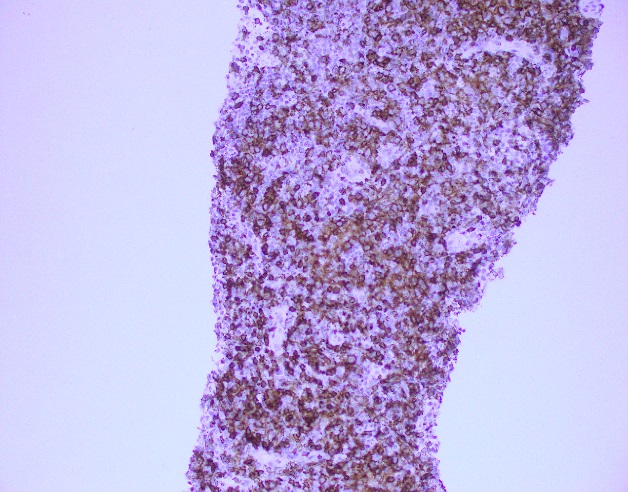
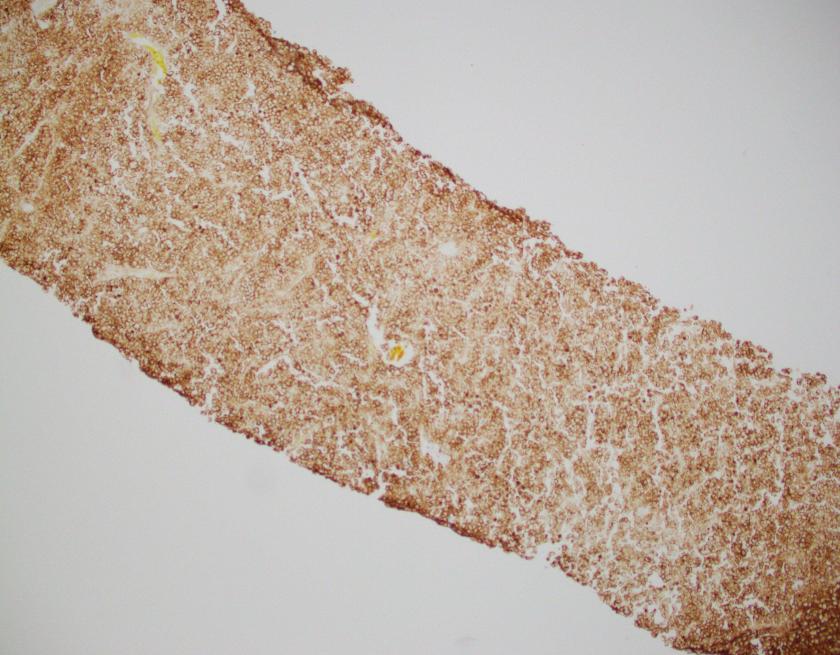
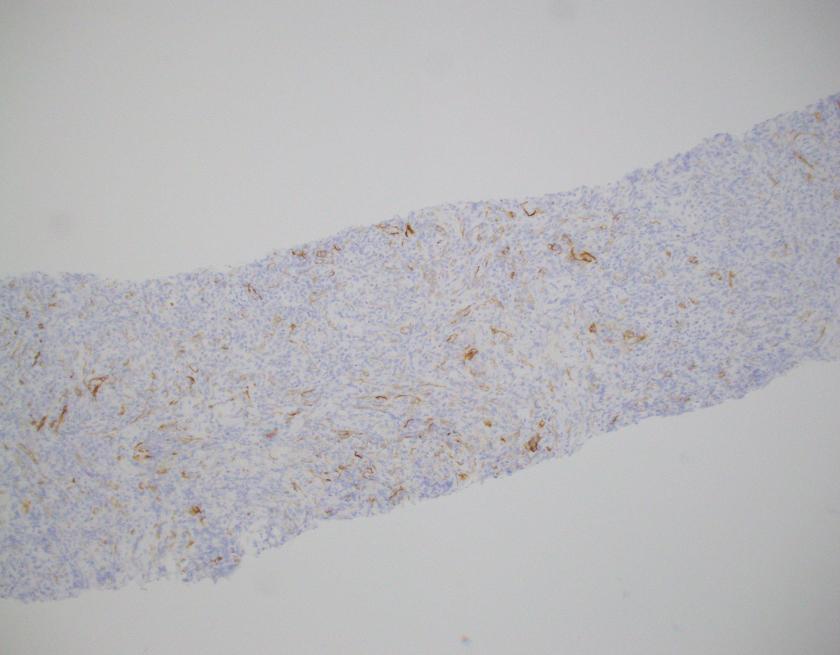
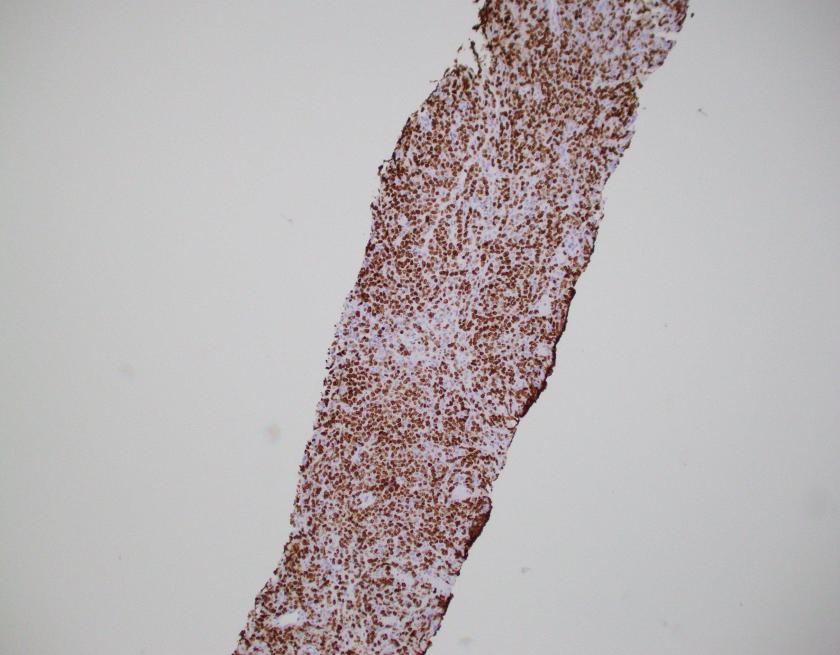
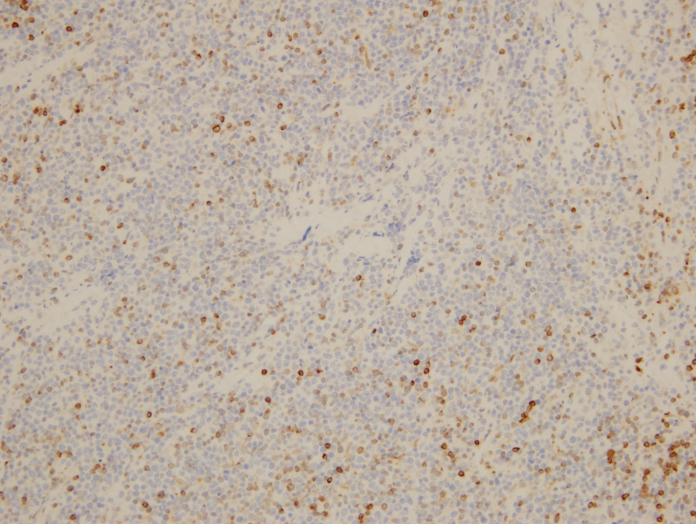
What about immunohistochemical staining for the histiocytes? While IHC can help outline histicoytic cells, unfortunately, quantitation of hemophagocytic histiocytes in the core biopsy or clot sections with the aid of CD68 immunostains does not correlate well with disease probability either.
Overall, the nonspecificity of hemophagocytosis in the marrow, even when present in high amounts, should remind both pathologists and clinicians that an isolated finding of hemophagocytosis lacks specificity and does not necessarily suggest HLH when the clinical presentation and laboratory findings are not compatible with the diagnosis. However, there still remains value in bone marrow biopsy examination in cases where clinical suspicion for HLH is high; in order to exclude other marrow processes; and in the rare case where there may not have been clinical suspicion of HLH but the presence of hemophagocytic histiocytes can raise that differential.
For additional images of hemophagocytic histiocytes check out these amazing picture tweets by Dr. Kate Dannheim (@KDannheimMD) https://twitter.com/KDannheimMD/status/933128799818002432) and Dr. Bharat Ramlal (@BeRaad87): https://twitter.com/HHPathology/status/926087105381531648
References
- Ho C, Yao X, Tian L, et al. Marrow Assessment for Hemophagocytic Lymphohistiocytosis Demonstrates Poor Correlation with Disease Probability. Am J Clin Pathol. 2014 Jan;141(1):62-71
- Hunter JI et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007 Feb;48(2):124-31.

-Ayse Irem Kilic is a 1st year anatomic and clinical pathology resident at Loyola University Medical Center. Follow Dr. Kilic on twitter @iremessa.

-Kamran M. Mirza, MD PhD is an Assistant Professor of Pathology and Medical Director of Molecular Pathology at Loyola University Medical Center. He was a top 5 honoree in ASCP’s Forty Under 40 2017. Follow Dr. Mirza on twitter @kmirza.