A 7 year old male with a history of restrictive cardiomyopathy status-post orthotopic heart transplant in June, 2010 that was on maintenance doses of tacrolimus and mycophenolate mofetil presented to his primary pediatrician left neck swelling. Starting in January 2017, the patient began with neck pain and swelling in the context of a recent gastrointestinal illness. Per CT report of the neck, a rim enhancing well-defined suppurative level III lymph node measuring 1.4 x 1.2 x 2.1 cm with adjacent soft tissue inflammatory changes extending into the left parapharyngeal space was identified. The patient was subsequently started on antibiotics and was discharged home with some improvement of swelling and pain.
The patient then presented again with continued neck swelling, although painless this time, and the patient’s cardiologist was contacted, who recommended a decrease in tacrolimus dosing. An otolaryngology evaluation was requested and given the concerning findings, the patient was admitted for further work-up, including a biopsy with a lymphoma protocol.



Results
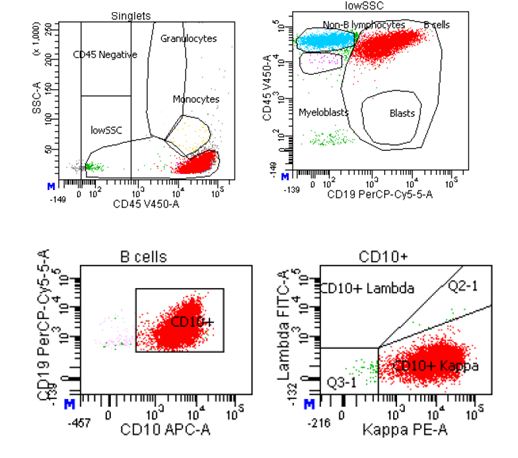
Flow cytometry revealed a kappa restricted CD10 positive mature B-cell population.
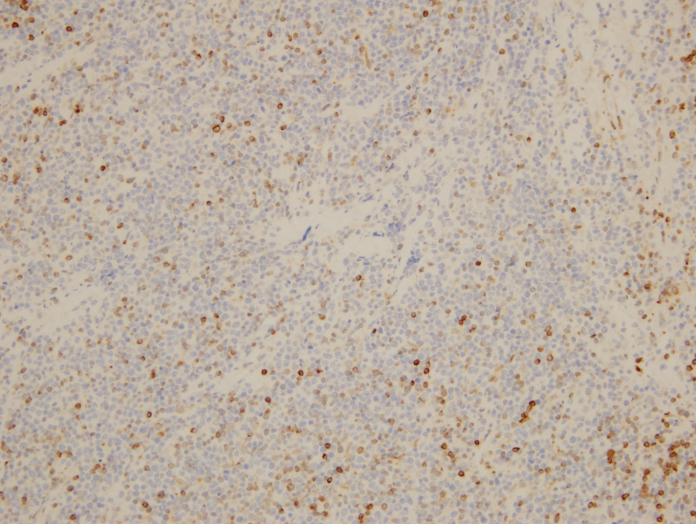
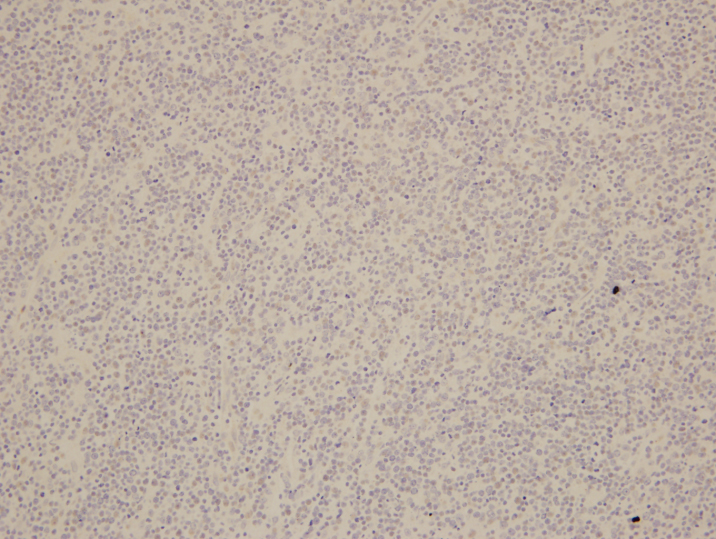
On biopsy examination, a population of monotonous lymphoid cells that are large in size with round to mildly irregular nuclear contours, open chromatin, and multiple inconspicuous nucleoli are present in a diffuse pattern. Abundant apoptotic bodies and mitotic figures are noted and occasional “starry sky” features are present. By immunohistochemistry, BCL6 highlights the neoplastic lymphocytes while BCL2 highlights background T-cells. EBER is negative.
Overall, despite a negative t(8;14) IGH/MYC translocation, the findings are best considered to be of an EBV-negative post-transplant lymphoproliferative disorder with morphologic features consistent with Burkitt lymphoma.
Discussion
Post-transplant lymphoproliferative disorders (PTLD) are a relatively rare complication in a variety of transplants that occurs in 2-10% of post-transplant patients. Overall, following a solid organ transplant (SOT), PTLD development is 1-5% of recipients with the highest incidence in intestinal and multivisceral transplantations (5-20%). Another factor is EBV status of the recipient, for which those that are EBV-naïve and lack cellular immunity to EBV are susceptible to graft-mediated EBV infection and ultimately developing an increased incidence in early PTLD. This population is overrepresented by pediatric transplant recipients1.
The presentation is highly variable and ranges from benign proliferations to overt lymphoproliferative disorders. Classifications for PTLD include early lesions, which are oligo- or polyclonal proliferations of EBV positive B cells have either a predominant infectious mononucleosis-like proliferation or a plasmacytic hyperplasia form. Polymorphic PTLD is a similar concept to the early proliferative lesions but the host architecture of the native structure is disrupted. Lastly, monomorphic PTLD is an entity that fulfills criteria for a non-Hodgkin lymphoma and is diagnosed according to the criteria of non-transplant associated lymphomas. Within pediatric registry studies, monomorphic PTLD accounts for 35-83% of all PTLD cases. B-cell lymphomas, particularly DLBCL, comprise the vast majority of monomorphic PTLD with plasmacytoma and T-cell lymphoproliferative disorders much less common2.
In this particular case, with the patient having been 7 years post-transplant and negative studies for EBV present, it is not surprising that germinal center phenotypic markers are highly expressed, such as CD10 and BCL6, which has been well elucidated by Jagadeesh, et al. Although not many genetic studies have been performed on post-transplant B-cell lymphomas, regardless of EBV status, there is some data demonstrating trisomies of 9 and/or 11 with translocations 8q24.1 (C-MYC), 3q24 (BCL6), and 14q32 (IGH). Rinaldi et al. noticed a lack of genetic lesions characteristic of postgerminal center derivation, such as gain of chromosome 3 (FOXP1, BCL6, and NFKBIZ) and 18q (BCL2 and NFATC1) together with losses of 6q (PRDM1 and TNFAIP3) in post-transplant DLBCL. A number of DNA mutations have also been described including genes associated with somatic hypermutation (SHM) such as PIM-1, PAX5, C-MYC, and RhoH/TTF. These particular mutations are also found to be independent of EBV status1.
Overall, post-transplant lymphoproliferative disorders occur in a variety of transplant settings across many age groups and can be dependent on EBV and CMV status as well as the type and degree of immunosuppression. Although many variations take place in PTLD, patients with the monomorphic type are diagnosed according to their non-transplant counterparts. Current perspective includes further analysis of molecular and cellular mechanisms incorporated into research projects, which could better aid in prognostic implications and future therapeutics.
- Morscio, et al. “Molecular pathogenesis of B-cell posttransplant lymphoproliferative disorder: What do we know so far?” Clinical and Developmental Immunology 2013.
- Mynarek, et al. “Posttransplant lymphoproliferative disease after pediatric solid organ transplantation,” Clinical and Developmental Immunology 2013.

-Phillip Michaels, MD is a board certified anatomic and clinical pathologist who is a current hematopathology fellow at Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA. His research interests include molecular profiling of diffuse large B-cell lymphoma as well as pathology resident education, especially in hematopathology and molecular genetic pathology.
