Case
A 100 year old female was seen for follow-up for her hypertension, mild renal impairment, and fatigue. The patient also stated a three week duration of pain in the area of the right upper quadrant that radiates to her back. No other symptoms or concerns were expressed.
An abdominal CT was performed which showed a 6.6 x 2.1 cm soft tissue mass in the right posterior chest wall that also encases the 11th rib. Given the concern for a malignant process, a core needle biopsy was obtained for histology only.
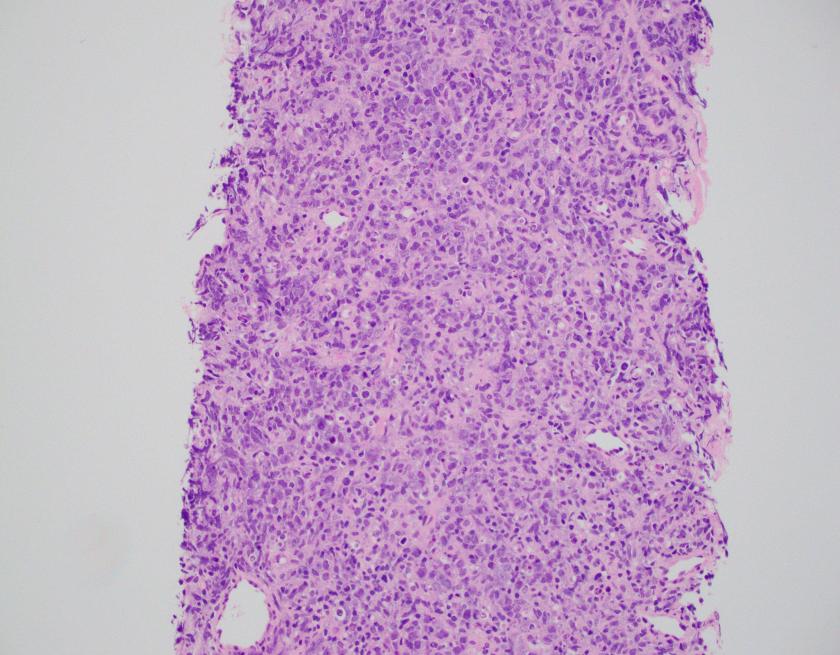
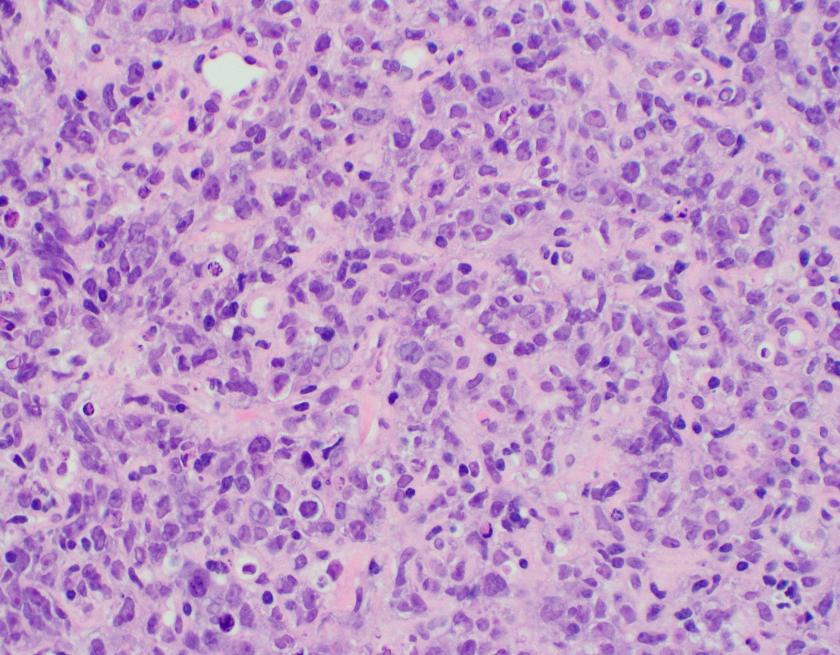
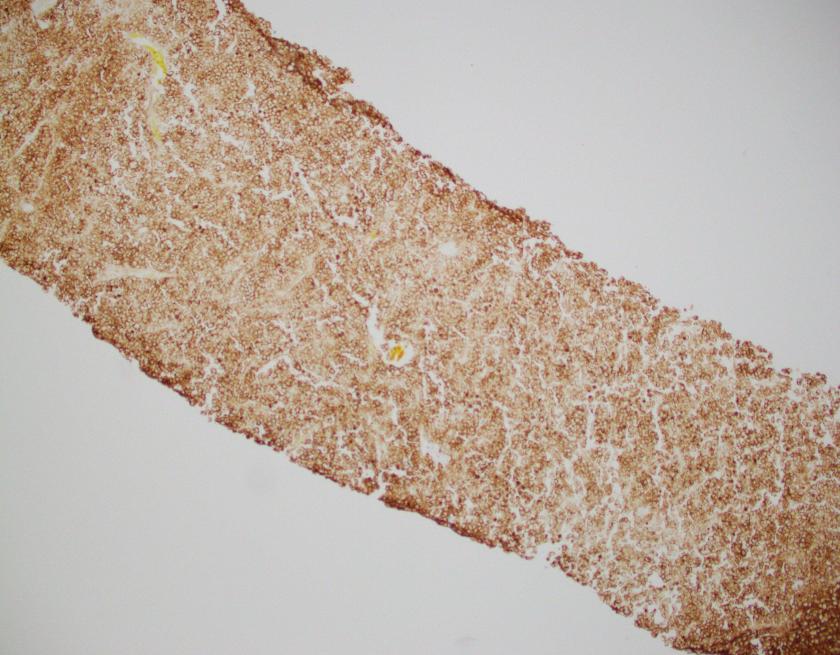
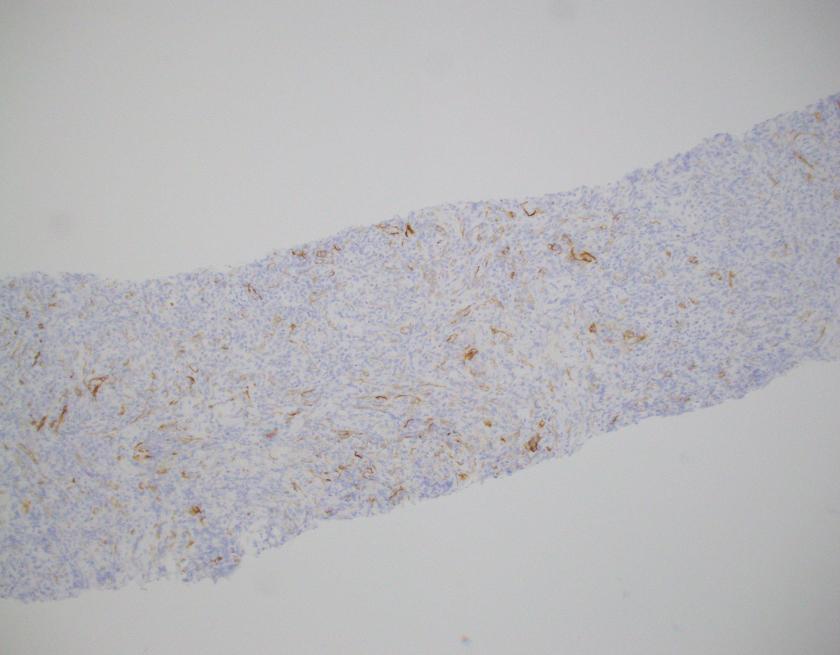


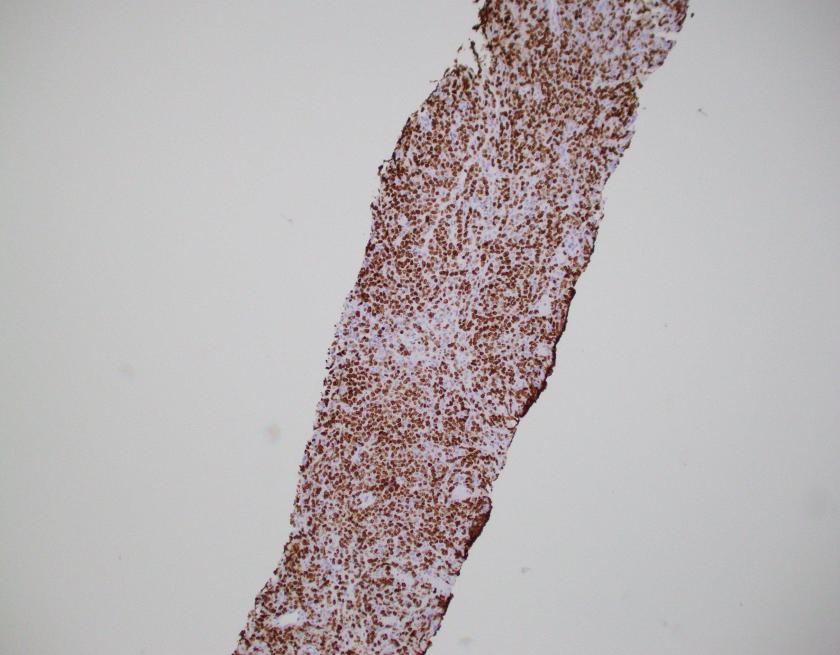
The H&E stained sections show a diffuse infiltration of atypical lymphoid cells that are large in size with irregular nuclear contours, vesicular chromatin, and some with prominent nucleoli. Frequent apoptotic bodies and mitotic figures were seen. By immunohistochemistry, CD20 highlights the infiltrating cells, which are positive for BCL2, BCL6, and MUM1 (major subset). CD10 is negative within the atypical lymphoid population. CD3 highlights background T-cells. Ki-67 proliferation index is approximately 70%. EBER ISH is negative.
Overall, the findings are consistent with diffuse large B-cell lymphoma, NOS with a non-GCB phenotype by the Hans algorithm.
Discussion
Diffuse large B-cell lymphoma (DLBCL) is the most common B-cell lymphoma in adults comprising 30%-40% of new adult lymphomas. Approximately 50% of patients will be cured, even in advanced cases; however, those that fail conventional therapy ultimately succumb to their illness.1 Up to 30% of patients have refractoriness or relapse after initial therapy with rituximab based regimens, particulary R-CHOP (ritixumab, cyclophosphamide, doxorubicin, vincristine, and prednisone).
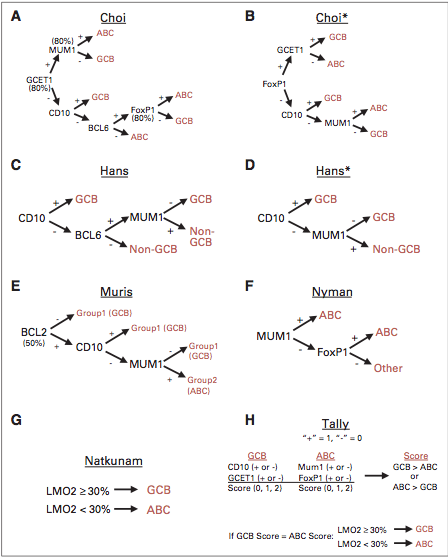
In the era of new molecular techniques and in the context of the heterogeneous nature of DLBCL, it has become important to accurately assess cell of origin (COO) as this has prognostic implications. With the seminal paper from Alizadeh and colleagues, gene expression profiling (GEP) by a microarray platform produced the concept of germinal center (GCB) versus activated B-cell (ABC) types of DLBCL.2 In the context of prognosis and R-CHOP therapy, the GCB type has a 3 year PFS of 75% as opposed to the ABC type that has a 3 year PFS of 40% (P<.001).3 Although GEP analysis is considered the ideal modality for determining COO, however, given the constraints of most modern hematopathology practices, surrogate immunohistochemical algorithms were developed to aid in COO determination. Of the multiple algorithms, the Hans algorithm is the most widely used and accepted for IHC determination of COO.
The COO determination has revealed multiple genetic alterations that are shared between the GCB and ABC phenotype while distinct changes have been identified in each type. Molecular mechanisms at play include, but are not limited to, histone modification, blocks to terminal differentiation, cell cycle activation, PI3K/AKT signaling activation, mTOR pathway activation, as well as a multitude of other signaling cascades. A common shared dysregulated pathway between GCB and ABC types include mutations in CREBBP and EP300, which is in approximately 30% of DLBCL cases and slightly enriched in the GCB group. Mutations/deletions in these genes result in inactivation and alter histone modification subsequently thought to contribute to acetylation of BCL6, which is a key regulatory protein in lymphomagenesis. Up to 33% of DLBCL have mutations in MLL2, which has a broad effect on chromatin regulation and epigenomic alteration. Approximately 35% of DLBCL cases with up to two- to three-fold increase in ABC type cases have genetic alterations in BCL6, particularly chromosomal rearrangements and mutations in the 5’ sequence. Pasqualucci et al also described other factors that lead to BCL6 inactivation, including mutations in MEF2B and FBXO11.4
ABC type DLBCL often displays canonical pathway activation of NF-ƙB signaling, which ultimately promotes survival, proliferation, and inhibition of apoptosis. This potentially is a result of alterations in the CBM signalosome (CARD11, BCL10, and MALT1) with up to 10% of ABC-DLBCL cases having a mutation in CARD11. Another modality of ABC activation is through the B-cell receptor signaling pathway in which 20% of cases harbor a CD79A or CD79B mutation. Interestingly enough, recurring mutations in MYD88 occur in ~30% of ABC-DLBCLs, which results in upregulation of NF-kB and Janus kinase-signal transducers. Other important genetic alterations include involvement by signaling pathways of spleen tyrosine kinase (SYK), PI3K, Bruton tyrosine kinase (BTK), and protein kinase C-β (PKC-β).
GCB type DLBCL often expresses CD10, LMO2, and BCL6 and has a less understood and distinct pathway when compared to ABC-DLBCL. The most common alterations include t(14;18) IGH-BCL2 (30-40%), C-REL amplification (30%), EZH2 (20%) and PTEN mutations (10%). These changes are almost never seen in ABC-DLBCL.

Although the findings in GCB and ABC type DLBCL are described, they are not absolute and multiple studies done by whole exome sequencing (WES) and whole genome sequencing (WGS) have elucidate further complexities and genetic changes. In 2015, data from Novak and colleagues revealed CNAs and mutations that were associated EFS, which also underscored the important 24 month milestone for survival.5 Morin et al in 2013 described 41 novel genes in DLBCL which demonstrated just how complex and heterogeneous DLBCL truly is (see figure below).6

As common as DLBCL is, there is much to be understood not only for lymphomagenesis, but for correct classification and risk stratification. Many targeted therapies have been designed and are in trials at the moment, but given the nature of DLBCL and its heterogeneity, more work on the molecular front is needed. Modalities for assessing COO are currently on the market but are not widely used. Perhaps COO determination by IHC may be an antiquated method, but it is currently the standard by which most pathologists practice. Overall, DLBCL in all its forms is not a uniform entity that can easily be defeated, but requires thought and diligence in achieving a cure.
- Lohr, JG et al. “Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing,” Proc Natl Acad Sci USA. 2012; 109(10): 3879-3884
- Alizadeh AA, et al. “Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling,” Nature 2000, 403:503-11
- Sehn, L and Gascoyne, R “Diffuse large B-cell lymphoma: optimizing outcome in the context of clinical and biologic heterogeneity,” Blood. 2015;125(1):22-32
- Pasqualucci, L and Dalla-Favera, Riccardo, “The Genetic Landscape of Diffuse Large B Cell Lymphoma,” Semin Hematol. 2015 April; 52(2): 67-76
- Novak, AJ et al. “Whole-exome analysis reveals novel somatic genomic alterations associated with outcome in immunochemotherapy-treated diffuse large B-cell lymphoma,” Blood Cancer Journal (2015) 5
- Morin, R et al. “Mutational and structural analysis of diffuse large B-cell lymphoma using whole-genome sequencing,” 2013;122(7):1256-1265

-Phillip Michaels, MD is a board certified anatomic and clinical pathologist who is a current hematopathology fellow at Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA. His research interests include molecular profiling of diffuse large B-cell lymphoma as well as pathology resident education, especially in hematopathology and molecular genetic pathology.

Thank you for sharing the case and findings. This a good way of teaching our students