Case History
A 12 year old female presented with thrombocytopenia. Previous platelet count performed at a different facility showed a platelet count of <100K. Patient signs show history of bruising, no history of trauma, intermittent epistaxis.
Family history shows no history of anemia or hypothyroidism from either parent. Incidental finding of hypothyroidism was revealed for this patient when laboratory testing was performed.
Laboratory results
DAT: Negative
PT 11.7/INR 1.1
PTT 38.3
Platelet aggregation studies: Decreased response to ADP-Collagen-Epinephrine and Arachidonic Acid. Results of which are consistent with platelet dysfunction due to storage pool defect.
vonWillberand panel shows within range results for Factor 8, vW antigen and vW Ristocetin.
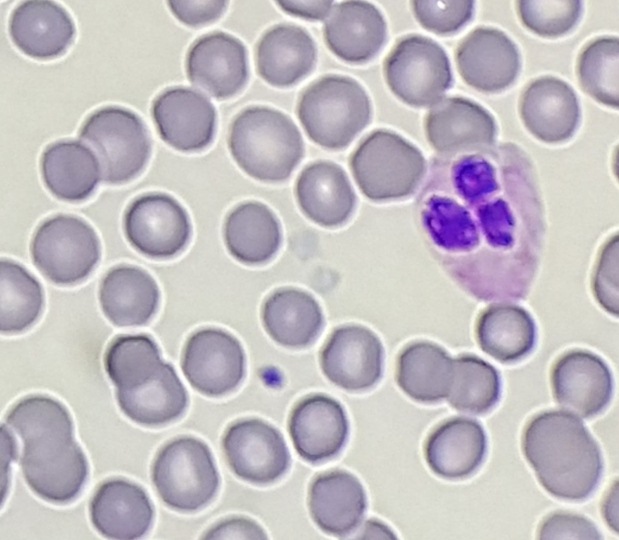
Peripheral blood smear shows light staining (gray) appearance of platelets.
Diagnosis: Gray Platelet Syndrome
Discussion
Gray platelet syndrome (GPS) is an inherited platelet disorder that presents with thrombocytopenia and characteristic pale/gray appearance of platelets under light microscopy. This gray appearance of platelets is due to the absence of alpha granules and their constituents.
According to Gunay-Aygun et al., the diagnosis of GPS requires demonstration of the absence or marked reduction of α-granules in platelets observed by electron microscopy (EM). Megakaryocytes also show decreased α-granules. Platelet dense bodies and lysosomes are unaffected. Alpha granules, the most abundant vesicles in platelets, store proteins that promote platelet adhesiveness and wound healing when secreted during platelet activation. Some α-granule proteins (eg, platelet factor 4 and β-thromboglobulin) are synthesized in megakaryocytes and packed into the vesicles, whereas others are either passively (eg, immunoglobulins and albumin) or actively (eg, fibrinogen) taken up from the plasma by receptor-mediated endocytosis. Proteins synthesized in megakaryocytes are markedly reduced in GPS, whereas other α-granule constituents are less affected. Studies of granule membrane-specific proteins have shown that platelets and megakaryocytes of GPS patients have rudimentary α-granule precursors. Therefore, the basic defect in GPS is thought to be the inability of megakaryocytes to pack endogeneously synthesized secretory proteins into developing α-granules. (Gunay-Aygun et al, 2010).
Most patients who present with GPS are characteristically macrothrombocytopenic and the number of megakaryocytes in the bone marrow appears normal. However platelet survival is reduced. This inability of megakaryocytes to survive is due to the alpha granule deficiency of this disorder therefore leading to thrombocytopenia. Myelofibrosis and splenomegaly is also apparent on patients with GPS but severe hemorrhage is unlikely, bleeding tendencies tend to be mild to moderate for GPS.
Most patients had bleeding symptoms from infancy with the average onset of 2 years of age. Average age of diagnosis is 10-14 years of age; some patients who have Gray Platelet Syndrome have presented with initial diagnosis of ITP (idiopathic thrombocytopenic purpura).
Reference
Gunay-Aygun, M., Zivony-Elboum, Y., Gumruk, F., Geiger, D., Cetin, M., Khayat, M., . . . Falik-Zaccai, T. (2010). Gray platelet syndrome: natural history of a large patient cohort and locus assignment to chromosome 3p. Blood, 116(23), 4990-5001. doi:10.1182/blood-2010-05-286534
-Written in collaboration with Stephanie Foster, BS MLS

-Carlo Ledesma, MS, SH(ASCP)CM MT(ASCPi) MT(AMT) is the program director for the Medical Laboratory Technology and Phlebotomy at Rose State College in Midwest City, Oklahoma as well as a technical consultant for Royal Laboratory Services. Carlo has worked in several areas of the laboratory including microbiology and hematology before becoming a laboratory manager and program director.
