Case History
We were asked to review the peripheral blood smear of an 18-year-old male who had presented to the emergency department with shortness of breath and abdominal distension. His past medical history was significant for numerous hospitalizations for recurrent fungal and bacterial pneumonia, pulmonary abscesses, osteomyelitis, necrotizing granulomas, and cervical lymphadenopathy requiring multiple lymphadenectomies. This history dates back to when he was 3 months old.
Blood Smear findings
The CBC demonstrated severe anemia and mild leukopenia. The peripheral blood smear showed numerous acanthocytes and poikilocytosis shown below.


Additional Clinical Findings
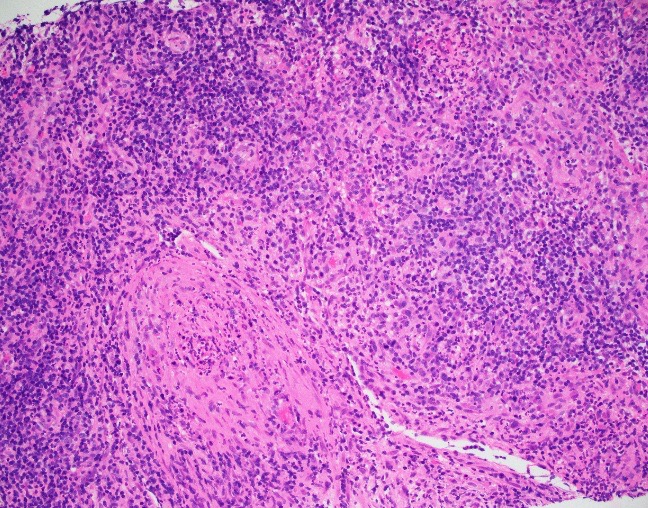
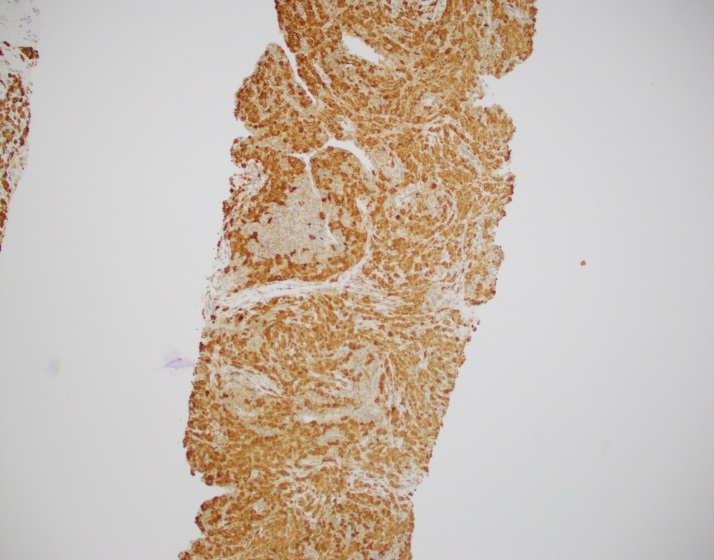
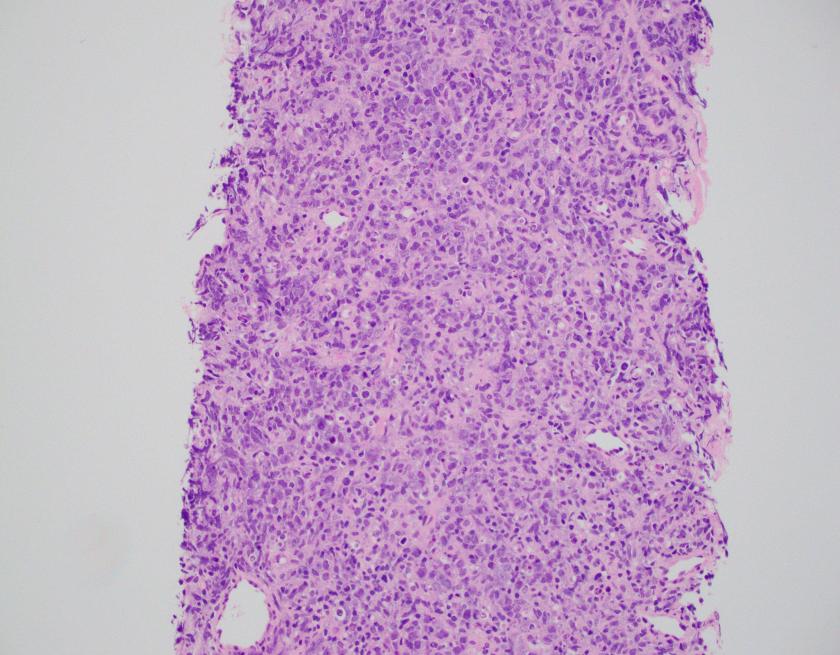
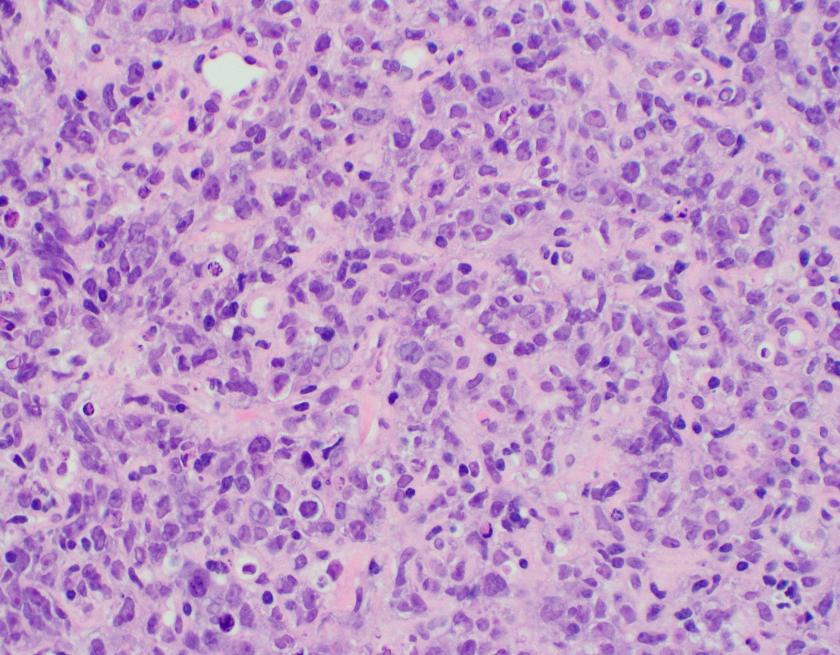
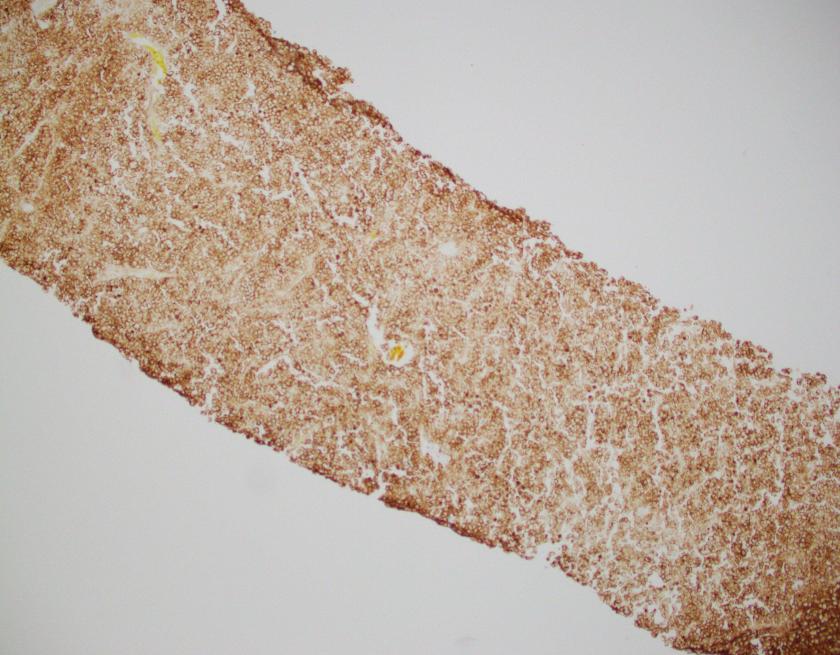
Abdominal ultrasonography demonstrated hepatosplenomegaly with enlarged porta-hepatis lymph nodes. Additionally, chest CT scanning demonstrated bilateral mass-like consolidations, prominent hilar lymphadenopathy, and osteolytic lesions of the vertebral bodies. A comprehensive investigation for opportunistic infections was negative. Lung and vertebral body biopsies (not pictured here) revealed poorly formed granulomas. A blood transfusion was considered; however, the patient had previously been demonstrated to express anti-Kx antibodies, which would require transfusion with exceedingly rare blood products.
Diagnosis
The preceding case history describes a patient with a contiguous gene deletion syndrome that includes chronic granulomatous disease (CGD) and the McLeod phenotype, demonstrating a fascinating disorder with important implications in hematopathology and several other disciplines of pathology.
Discussion
McLeod syndrome is a rare, X-linked disorder characterized by the deletion of the XK gene which encodes for the Xk protein. Overall, the lack of synthesis of the Xk protein leads to the lack of expression of the Kx antigen which in turn leads to a marked decrease in the quantities of Kell antigens. In this case, due to the presence of an anti-Kx antibody, the patient would require transfusion with either Kell-null or McLeod phenotype blood products. Unfortunately, only one unit of compatible blood was identified when the rare blood donor database was queried. The clinical team therefore elected for treatment with erythropoietin and iron supplementation which eventually lead to a modest increase in the patient’s hemoglobin concentration.
Acanthocytes, or spur cells, are spiculated red cells with a few projections of varying size and surface distribution that can be seen in a variety of clinical conditions including CGD with McLeod red cell phenotype. Other conditions include (but are not limited to) neuroacanthocytosis, malnutrition states, infantile pyknocytosis, (Lu) null Lutheran phenotype, hypothyroidism, myxedema, and Zieve syndrome. Acanthocytes should be distinguished from echinocytes, or burr cells, that also demonstrate multiple small projections but these are uniformly distributed on the red cell surface.
The prominent acanthocytosis seen in McLeod syndrome is thought to be due to an imbalance of the number of lipids in the inner layer relative to the outer layer. Related to this phenomenon is McLeod neuroacanthocytosis syndrome, a disorder with neurologic manifestations including movement disorders, cognitive alterations, and psychiatric symptoms. Although our patient did not exhibit these symptoms, McLeod neuroacanthocytosis syndrome is known to start in early to middle adulthood and the patient will need to be monitored for the onset of neurologic sequelae.
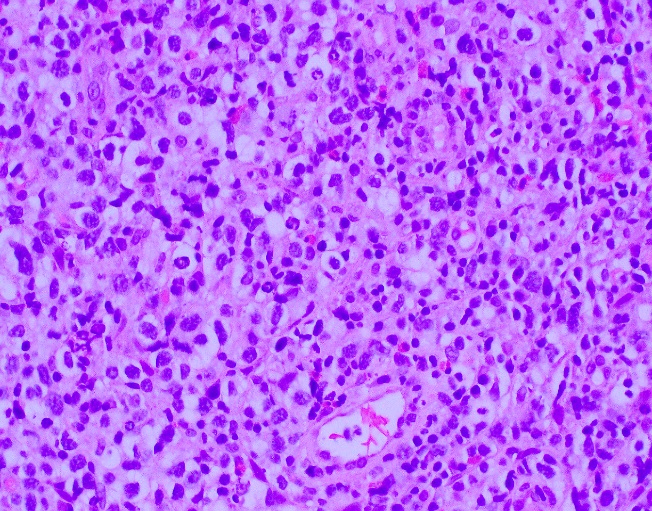
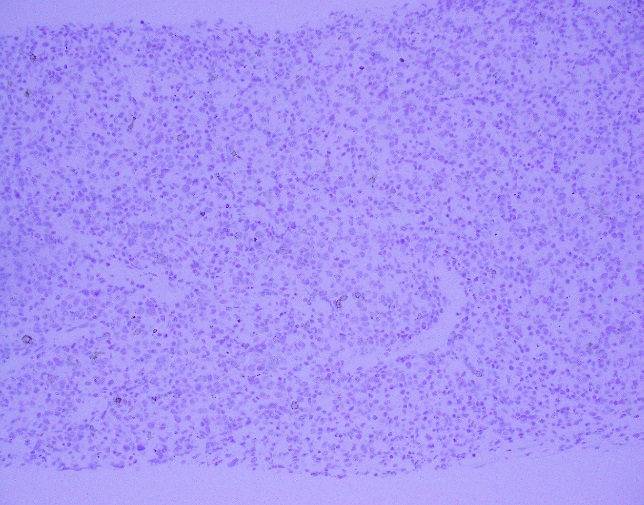
The McLeod phenotype is frequently associated with CGD due to the proximity of the XK gene to the CYBB gene on the X chromosome. The CYBB gene encodes for a subunit of the NADPH oxidase enzyme complex. A deficiency in NADPH oxidase activity leads to the characteristic increased susceptibility to severe bacterial and fungal infections seen in CGD. The nitroblue-tetrazolium test can be used to evaluate NADPH oxidase activity in the white blood cells and can help make a diagnosis of CGD. Histologically, CGD can show prominent necrotizing and non-necrotizing granulomas in various locations throughout the body.
Overall, treatment of CGD with McLeod red cell phenotype is supportive. There is no known cure or definitive treatment. The patient will likely continue to have infections with opportunistic organisms which will be treated on a case by case basis.
References
- Heyworth PG, Cross AR, Curnutte JT. Chronic granulomatous disease. Current opinion in immunology. 2003 Oct 31;15(5):578-84.
- Jung HH, Danek A, Walker RH, Frey BM, Gassner C. McLeod neuroacanthocytosis syndrome.
- Khodadad JK, Weinstein RS, Marsh LW, Steck TL. Shape determinants of McLeod acanthocytes. Journal of Membrane Biology. 1989 Mar 1;107(3):213-8.
- Watkins CE, Litchfield J, Song E, Jaishankar GB, Misra N, Holla N, Duffourc M, Krishnaswamy G. Chronic granulomatous disease, the McLeod phenotype and the contiguous gene deletion syndrome-a review. Clinical and Molecular Allergy. 2011 Nov 23;9(1):13.

-Michael Moravek, MD is a 2nd year anatomic and clinical pathology resident at Loyola University Medical Center. Follow Dr. Moravek on twitter @MoravekMD

-Kamran M. Mirza, MD PhD is an Assistant Professor of Pathology and Medical Director of Molecular Pathology at Loyola University Medical Center. He was a top 5 honoree in ASCP’s Forty Under 40 2017. Follow Dr. Mirza on twitter @kmirza.