Case History
A 69 year old female with a past medical history significant for endometrial adenocarcinoma, traumatic brain injury, atrial fibrillation, hypertension, hyperlipidemia, and persistent monocytosis (absolute monocyte count ranging from 1.6-3.7 K/uL) who had an indeterminate lesion identified in the T5 vertebra, read as “hemangioma, although surrounding edema is worrisome for malignancy” upon staging imaging for history of endometrial carcinoma.
CBC at the time was: WBC 9.8; HGB 12.9; HCT 37.6; PLT 154; MCV 92 fL; MCH 31.7.
Automated differential showed: 43.0 Neutrophils; 34.8 Lymphocytes; 20.0 Monocytes; 1.4 Eosinophils; 0.2 Basophils; 0.6 Immature granulocytes.
Absolute monocyte count was 1.95 K/uL.
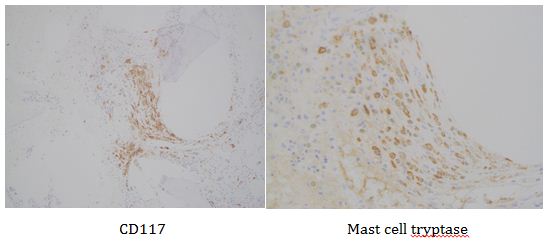
This lesion was biopsied and given the findings, a subsequent bone marrow biopsy was performed on 5/4/2017. The bone marrow core biopsy contained multiple compact aggregates of spindle shaped cells with hypogranular cytoplasm, morphologically compatible with atypical mast cells. Within these aggregates, numerous eosinophils are present. By immunohistochemistry, the mast cells are brightly positive for CD117 and mast cell tryptase. Concurrent bone marrow aspirate flow cytometry demonstrated a small population of mast cells that co-expressed CD2 and CD25.
Of note, the patient was found to have a persistent absolute monocytosis. Flow cytometry revealed an abnormal population of monocytes that displayed aberrant phenotypic expression of CD2 and CD56 (subset).
Next-generation sequencing revealed two truncation mutations in the TET2 gene (K988* in 34.6% of the reads and Q1138* in 36.4% of the reads). Cytogenetic analysis revealed a normal female karyotype (46,XX).


Overall, an immunophenotypically abnormal population of mast cells and monocytes are present in the context of a long-standing absolute monocytosis and the presence of two TET2 truncating mutations, supporting a diagnosis of systemic mastocytosis with an associated hematologic non-mast cell lineage disorder (best classified as chronic myelomonocytic leukemia).
Discussion
A diagnosis of systemic mastocytosis is a combination of clinical, morphologic, immunophenotypic, and molecular analyses, as required by the World Health Organization (WHO 2008). By current consensus guidelines, SM variants are partly distinguished by clinicopathologic criteria referred collectively as B and C findings. B findings include: >30% of bone marrow mast cells (MC) on biopsy and/or serum tryptase levels >200 ng/mL; increased marrow cellularity/dysplasia without meeting diagnostic criteria for another myeloid neoplasm; or enlargement of liver, spleen, or lymph nodes without evidence of organ damage. C findings include: evidence of organ damage caused by a local MC infiltrate, such as abnormal liver function and/or ascites, hypersplenism, cytopenias, large osteolytic lesions/fractures, and malabsorption with weight loss caused by MC infiltrate in the gastrointestinal tract.
Systemic mastocytosis commonly occurs in two types with different clinical courses based upon the aforementioned findings. Indolent SM (ISM) is defined by the absence of C findings. Smoldering SM is a subtype of ISM that displays 2 or more B findings. ISM may become more aggressive and a descriptive term of advanced SM refers to a category including aggressive SM (ASM), mast cell leukemia (MCL), and “SM with an associated myeloid neoplasm.” The latter entity comprises more than 90% of cases that have previously been referred to as SM with an associated hematologic non-mast cell lineage disorder (SH-AHNMD).
ASM and MCL are characterized by organ damage and histologic characteristics. ASM often exhibits multifocal bone marrow infiltration of atypical mast cells that are often spindled in shape with hypogranular or immature morphology. Marked fibrosis often accompanies the infiltrate as well as a KIT D816V mutation. MCL is codified by more than 20% of the marrow aspirate nucleated cells represent by mast cells and on core biopsy, a compact infiltrate is often identified with usually low level fibrosis. In MCL, circulating mast cells are greater than 10% of nucleated cells but according to Gotlib et al., the aleukemic MCL (less than 10% circulating mast cells) is more common.
In the context of our patient, myeloid neoplasms associated with SM are often represented by MDS, MPN, or MDS/MPN overlap disorders, and occasionally AML.
Associated lymphoid or plasma cell neoplasms have been described, but in a much lower frequency.
In accordance with the diagnostic implications, KIT D816V mutational analysis is important therapeutically. Most patients with SM harbor the KIT D816V mutation (>80% in one clinical series; 90-100% in research studies using purified MCs), which is a considered imatinib-resistant mutation. Midostaurin (a second generation TKI) may provide some disease response while nilotinib or dasatinib are usually less likely to lead to a durable response. The rare patients who have a juxtamembrane domain KIT mutation are much more likely to respond to imatinib or masitinib.
For disease response, criteria were first published in 2003 by Valent, et al. In a reiterated version published in 2007, the evaluation of clinical evidence of organ damage (C findings), was the foundation for determining appropriate response. Another facet to determining response was in relation to BM MC burden, serum tryptase level, and organomegaly, which further subcategorized the levels of major response (MR). MR was defined as normalization of 1 or more C findings. In turn, MR was divided into 3 categories:
- Complete remission (resolution of MC infiltrates in organs, serum tryptase less than 20 ng/mL, and disappearance of SM-associated organomegaly)
- Incomplete remission (decrease in MC infiltrates in organs and/or serum tryptase levels and/or visible regression of organomegaly by >50%)
- Pure clinical response (without decrease in MC infiltrates, serum tryptase levels, or organomegaly)
Partial response (PR) is defined as incomplete regression of 1 or more C findings and include good partial response (GPR; >50% regression of 1 or more C findings) and minor response (<50% regression).
Lastly, the Mayo Clinic published revised response criteria in 2010 which established minimal baseline laboratory abnormalities for organ damage to be evaluated in order to allow for more accurate assessment of response to therapy that is clinically more relevant.
Overall, systemic mastocytosis is a rare entity that displays a range of presentations that can be described as indolent up to an aggressive (advanced) phenotype. The hallmarks for diagnosis include histologic, immunophenotypic, molecular, and clinical findings.
References
- Gotlib, J et al. “International Working Group-Myeloproliferative Neoplasms Research and Treatment (IWG-MRT) & European Competence Network on Mastocytosis (ECNM) consensus response criteria in advanced systemic mastocytosis,” Blood, 2012.
- Horny HP et al. “Mastocytosis,” In: Swerdlow S et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC Press; 2008:53-63
- Valent P et al. “Aggressive systemic mastocytosis and related mast cell disorders: current treatment options and proposed response criteria.” Leuk Res. 2003;27(7):635-641.
- Pardanani A, et al. “A critical reappraisal of treatment response criteria in systemic mastocytosis and a proposal for revisions. Eur J Haematol. 2010;84(5):371-378.

-Phillip Michaels, MD is a board certified anatomic and clinical pathologist who is a current hematopathology fellow at Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA. His research interests include molecular profiling of diffuse large B-cell lymphoma as well as pathology resident education, especially in hematopathology and molecular genetic pathology.
