History
A 67 year old female presents with a two-month history of sore throat. She endorses dysphagia and left-sided otalgia but denies voice changes, shortness of breath, hemoptysis, weight loss, fever or night sweats. She has smoked 1 pack/day for 41 years and occasionally drinks alcohol. Her past medical history is notable for systemic lupus erythematosus for which she takes Plaquenil.
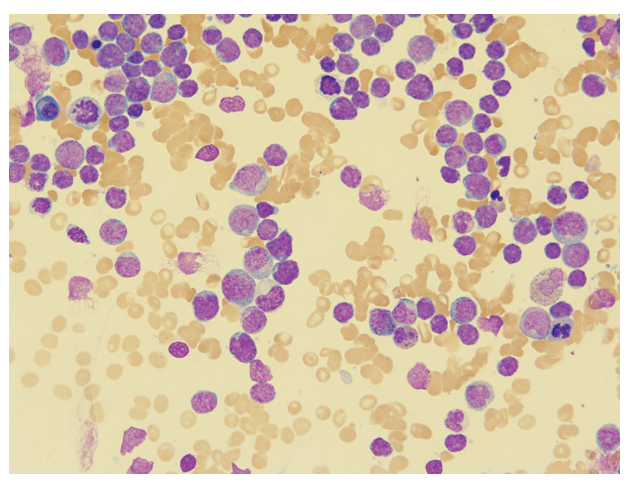
Physical examination slightly elevated systolic blood pressure. She is afebrile. Pertinent neck exam findings include mild tonsillar asymmetry (left slightly larger than right), and a firm mass at left base of tongue, and a 3 cm lymph node in the neck (left level III). A biopsy sample was taken from the tongue mass.
Biopsy
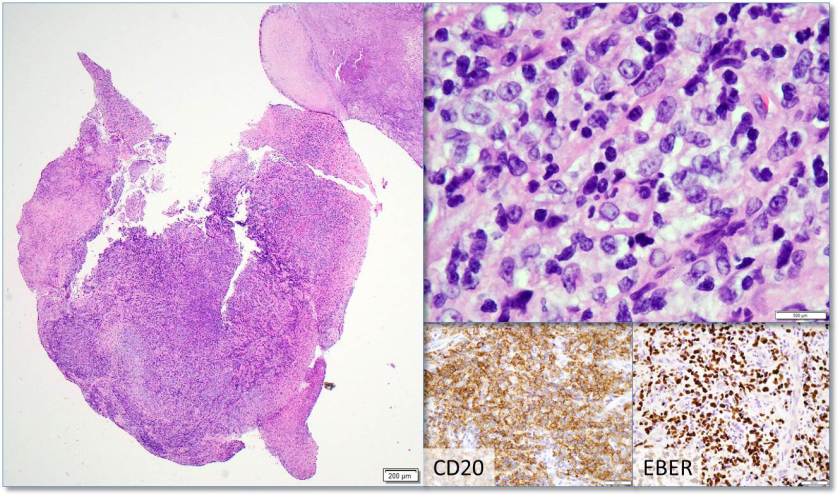
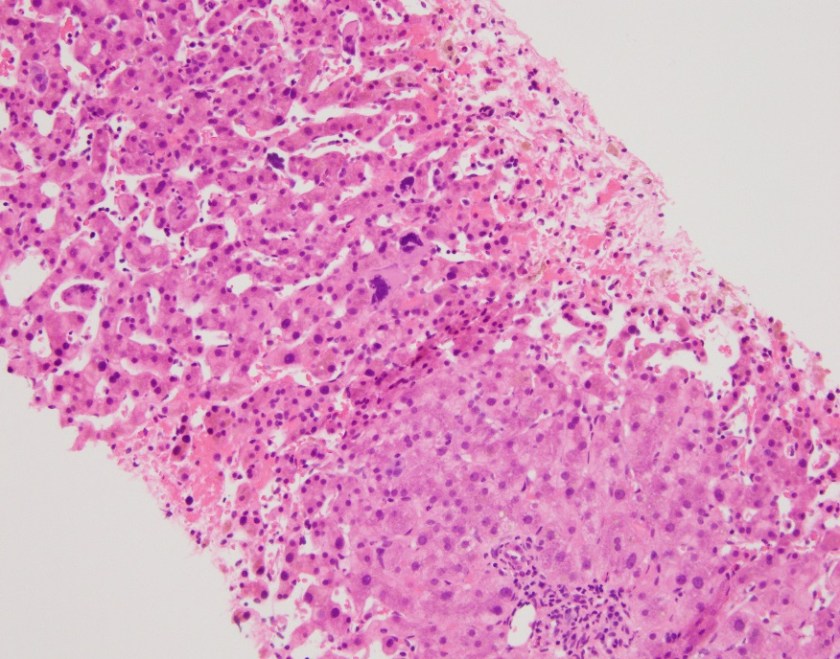
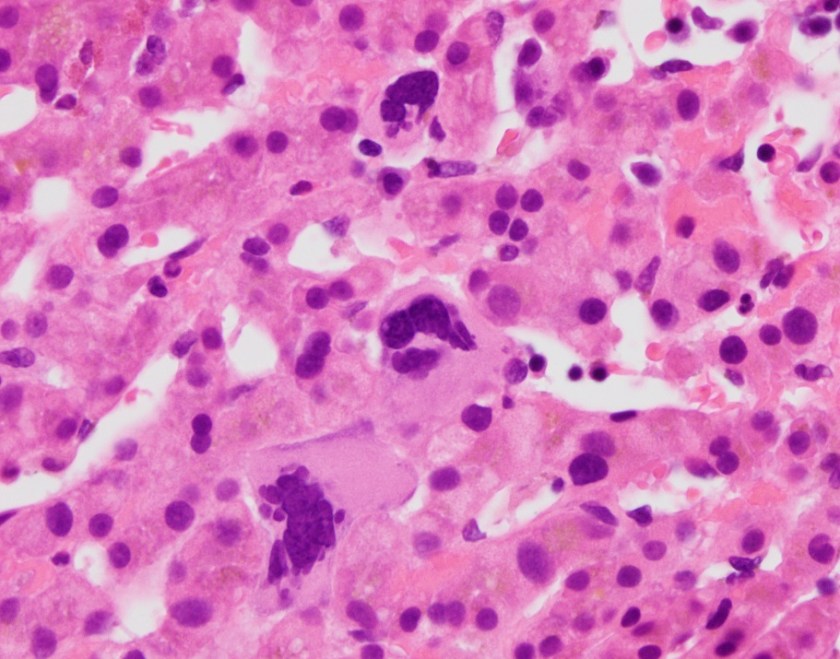
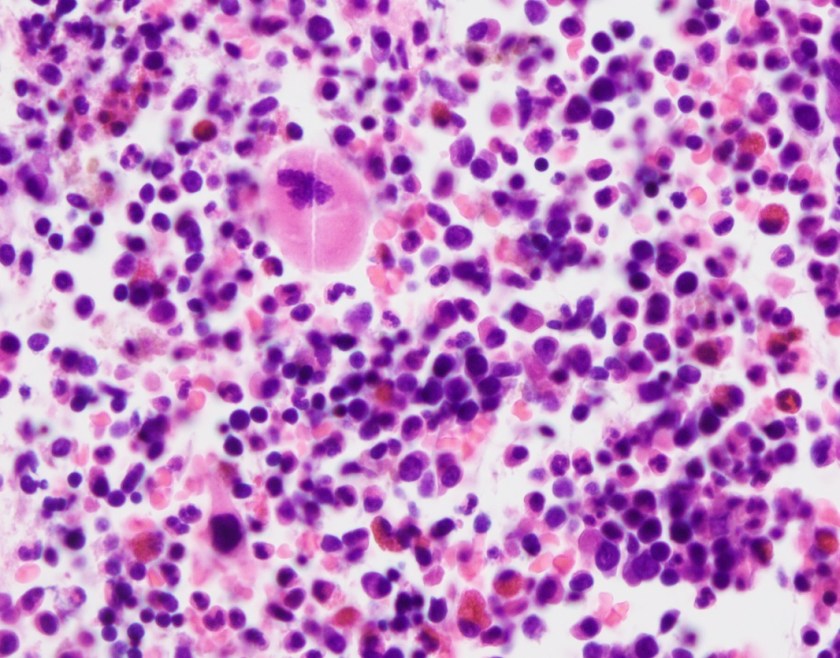
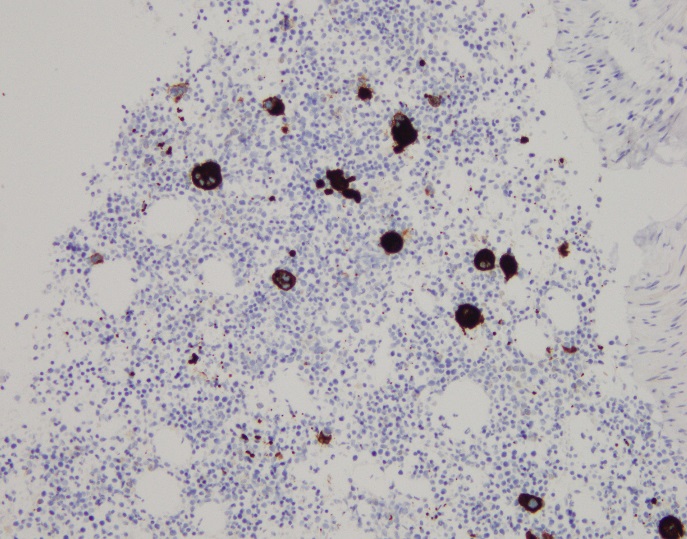
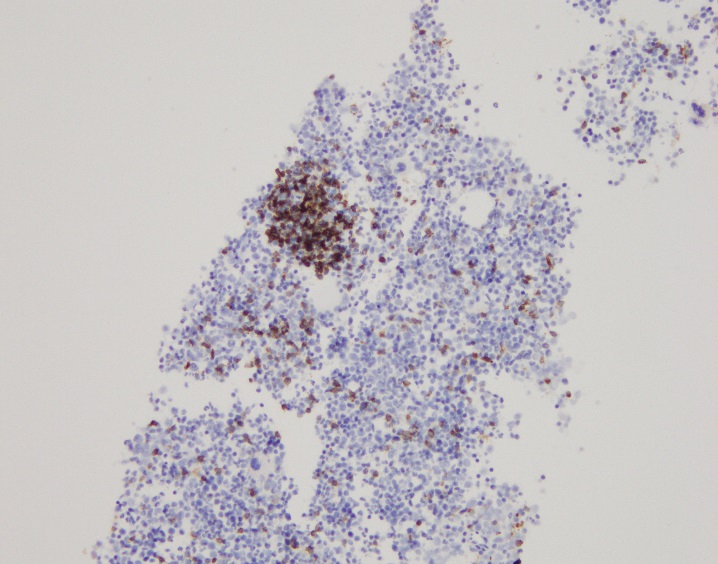
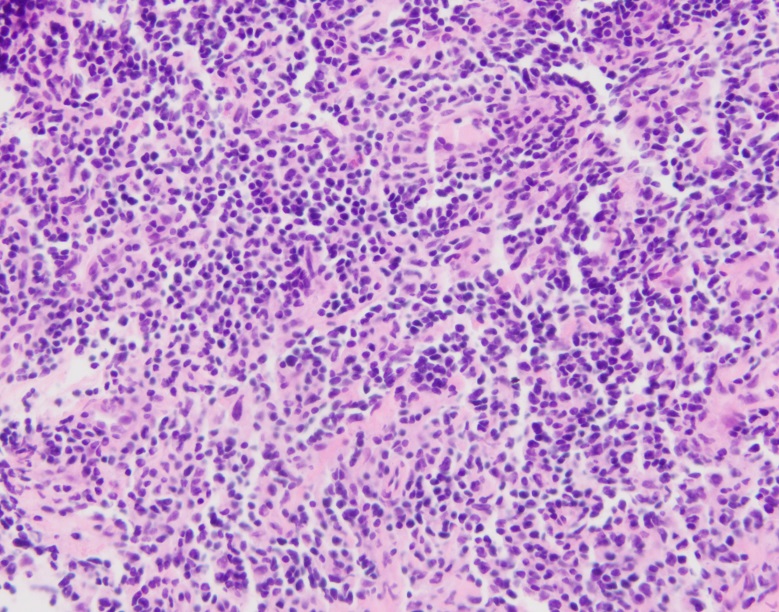
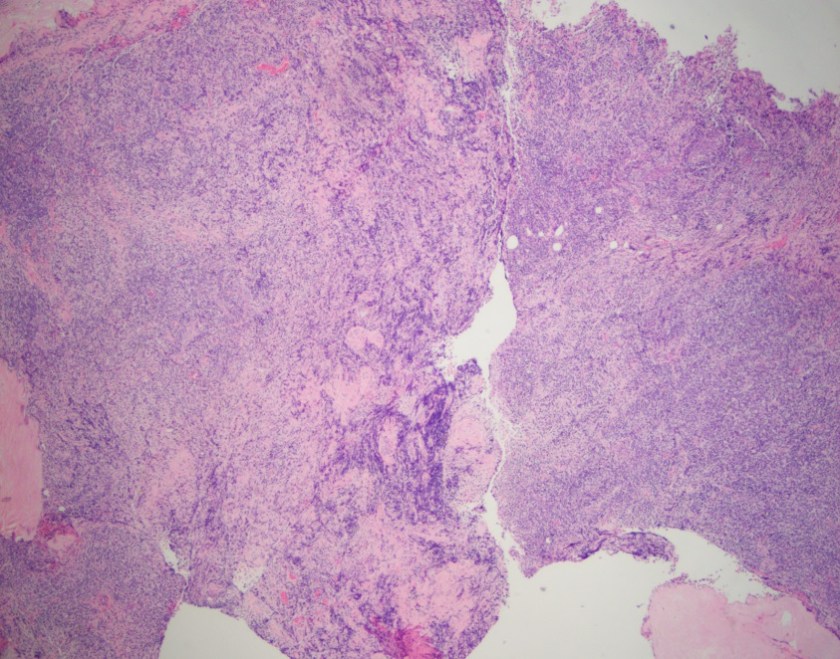
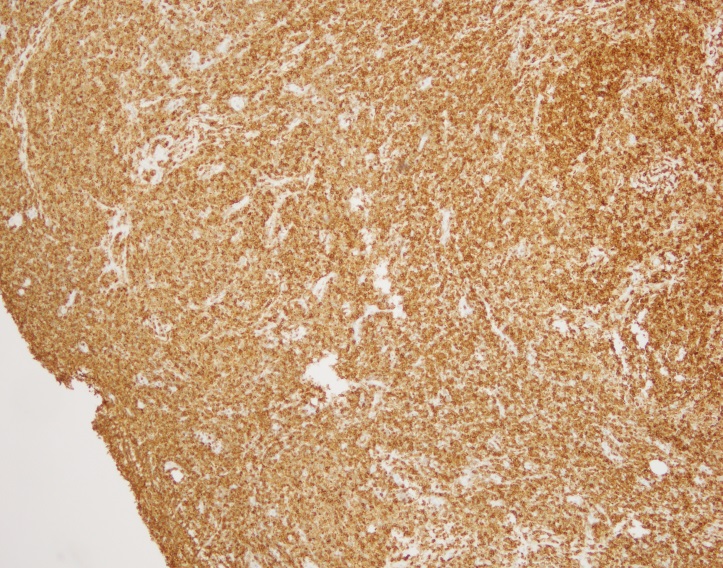
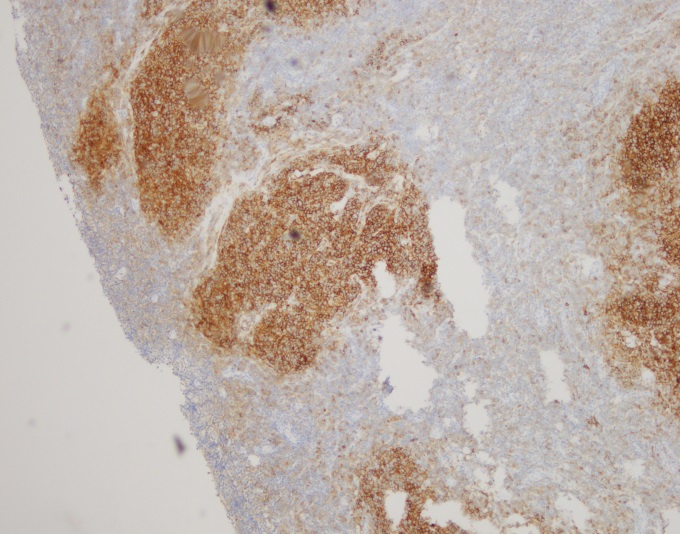
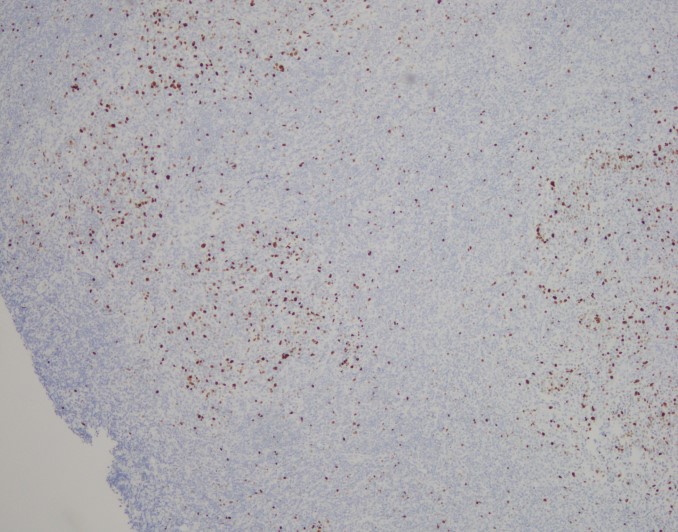
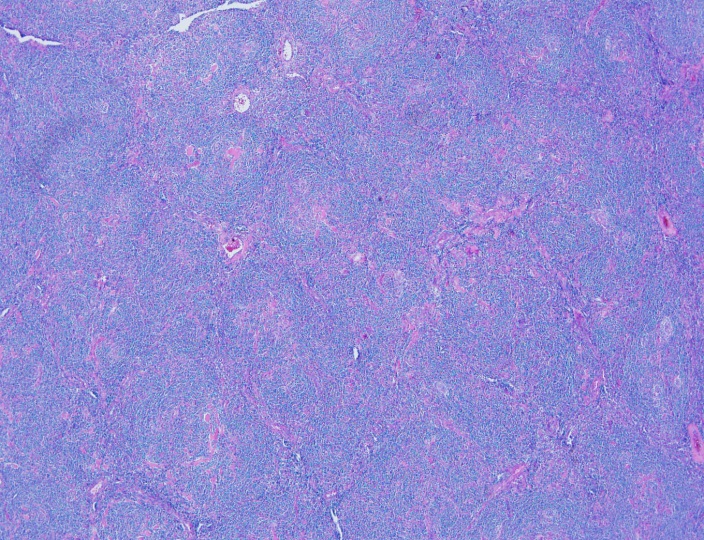
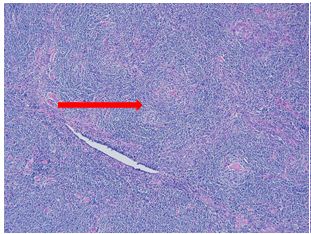
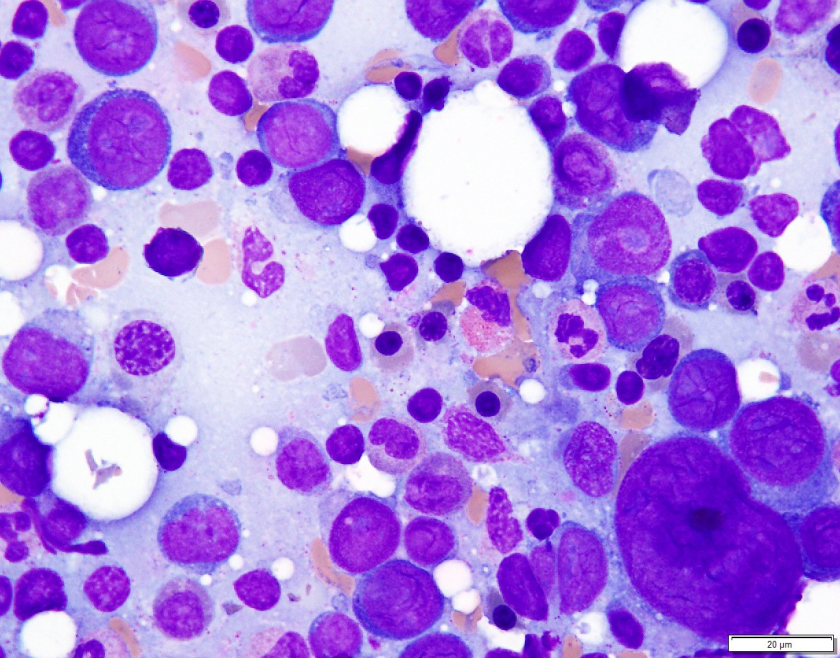
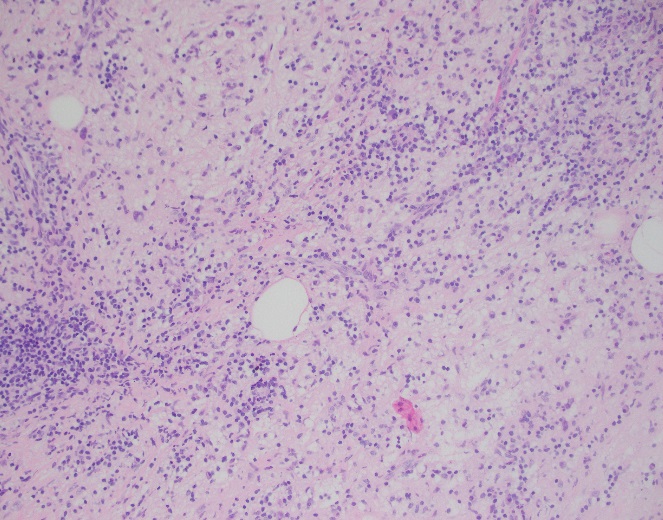
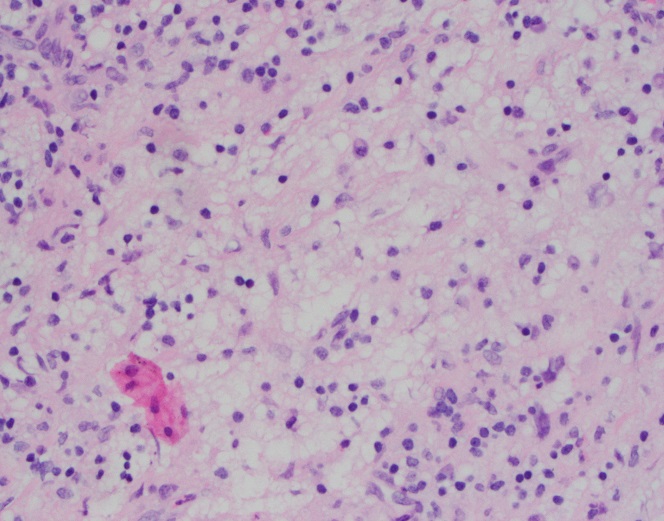
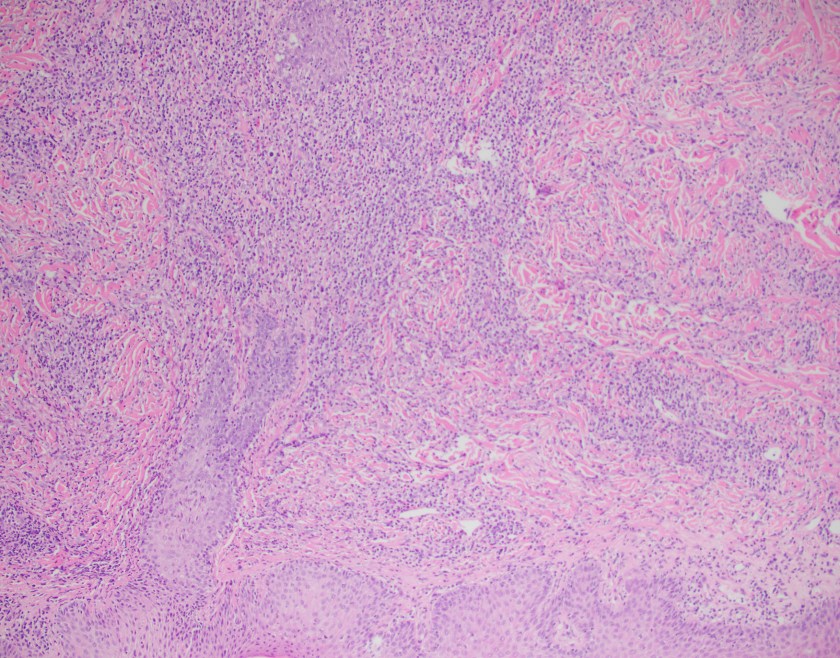
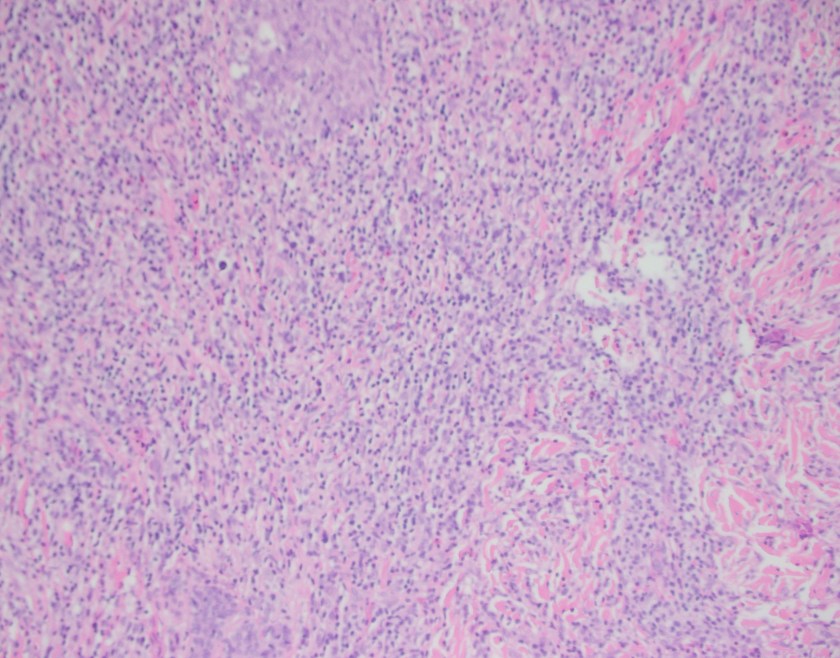
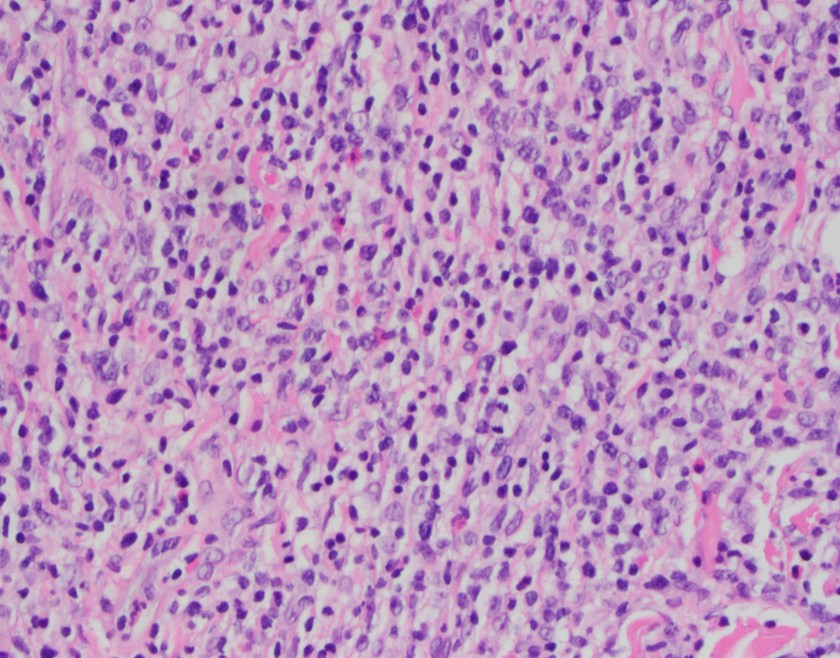
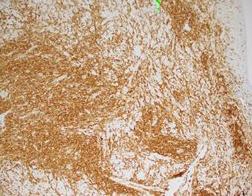
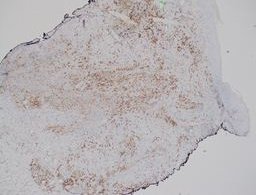
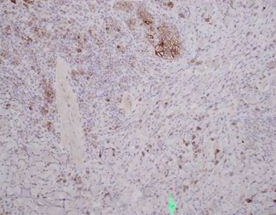
H&E stained sections reveal sheets of large lymphocytes. The lymphoid cells are medium to large in size with irregular nuclear contours and prominent nuclei. Areas of necrosis are prominent. No specific areas of epithelial ulceration are noted. Immunophenotypic characterization of the larger cells reveals positivity for CD20, CD30, CD79a, PAX5, MUM1, Epstein Barr virus encoded RNA (EBER) and a variable Ki-67 proliferation index, which is up to 60-70% in the larger cells, but around 20-30% overall. Only rare cells are positive for BCL-2 and BCL-6. The lymphoma cells are negative for keratin AE1/AE3, CD10, CD4, CD8, CD21, CD23, CD7, CD5, Cyclin D1, CD68, CD56, and CD43. The background T cells express CD5 and CD7 and are a mixture of CD4 and CD8 with CD4 predominance.
We considered the diagnosis of EBV-positive mucocutaneous ulcer (a more indolent entity); however, the lack of history of an ulcer/ulceration and the presence of a mass-lesion (with additional adenopathy) does not support this diagnosis.
The findings are most consistent with EBV-positive DLBCL, NOS (WHO 2017), previously known as EBV positive DLBCL of the elderly (WHO 2008).
Discussion
Epstein Barr Virus, a member of the Herpesviridae family is mostly known for causing Infectious Mononucleosis. However, the ubiquitous virus which is present in about 90% of adults but often asymptomatic1, has a predilection for epithelial cells including B-cells.2 Incorporation of the viral genome and viral takeover of the cells proliferative machinery underlies the pathogenesis of any EBV-related disease/malignancy. It has been associated with a gastric carcinoma, fulminant hepatitis, undifferentiated nasopharyngeal carcinoma, and B cell, T cell and NK cell lymphomas3, including EBV+ diffuse large B-cell lymphoma, not otherwise specified (DLBCL-NOS).
EBV-positive diffuse large B-cell lymphoma, not otherwise specified (EBV+ DLBCL-NOS) was formerly known as EBV-positive diffuse large B-cell lymphoma (DLBCL) of the elderly. The WHO classification substituted “not otherwise specified” in place of “for the elderly” to reflect two things: 1) EBV is associated with other specific neoplastic Large B-Cell diseases such as lymphomatoid granulomatosis, and 2) EBV+DLBCL can affect younger individuals as well as the elderly. 2
EBV+DLBCL-NOS patients may occur in nodal or extranodal sites, with up to 40% presenting with extranodal sites at least in the early stages. Patients may be asymptomatic with or without B symptoms but usually, patients present with rapidly enlarging tumors at single or multinodal sites, as well as at extranodal sites. 4
The patient’s presentation with sore throat and the finding of neck mass with EBV-positive large B-cells associated with ulcer-like necrosis raises a differential diagnosis that ranges from reactive to malignant. Table 1 shows a comparison between three differential diagnoses: EBV+DLBCL-NOS; EBV-positive mucocutaneous ulcer; and infectious mononucleosis.
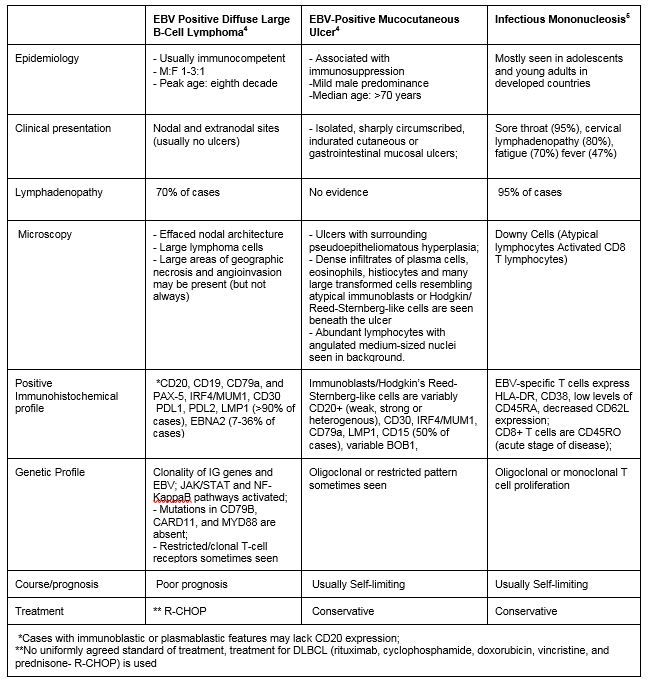
Unfortunately, there is currently no uniformly agreed standard of treatment for EBV+DLBCL which has a worse prognosis than EBV negative DLBCL.2 The standard treatment for DLBCL (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone- R-CHOP) is used but it responds poorly to treatment, with a median survival of 2 years.
Therefore, early detection by clinical suspicion and testing all DLBCL patients for EBV is very important.2
References
- Tsuchiya S. Diagnosis of Epstein–Barr virus-associated diseases. Critical Reviews in Oncology and Hematology. 2002;44(3):227-238. https://www.sciencedirect.com/science/article/pii/S1040842802001142. doi: 10.1016/S1040-8428(02)00114-2.
- Murthy SL, Hitchcock MA, Endicott-Yazdani T, Watson JT, Krause JR. Epstein-barr virus–positive diffuse large B-cell lymphoma. Proceedings (Baylor University.Medical Center). 2017;30(4):443-444. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC5595389/.
- Okano, Motohiko, MD, PhD|Gross, Thomas G., MD, PhD. Acute or chronic life-threatening diseases associated with epstein-barr virus infection. American Journal of the Medical Sciences, The. 2012;343(6):483-489. https://www.clinicalkey.es/playcontent/1-s2.0-S0002962915309435. doi: 10.1097/MAJ.0b013e318236e02d.
- Swerdlow S, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Arber D, Hasserjian R, Le Beau M. WHO classification of tumours of haematopoietic and lymphoid tissues. 2017.
- Dunmire SK, Hogquist KA, Balfour HH. Infectious Mononucleosis. Current topics in microbiology and immunology. 2015;390:211-240. doi:10.1007/978-3-319-22822-8_9.

-Adesola Akinyemi, M.D., MPH, recently earned his MPH-Health Policy and Management from New York Medical College. He plans on pursuing residency training in pathology. His interests include cytopathology, neuropathology, and health outcomes improvement through systems thinking and design.

-Kamran M. Mirza, MD PhD is an Assistant Professor of Pathology and Medical Director of Molecular Pathology at Loyola University Medical Center. He was a top 5 honoree in ASCP’s Forty Under 40 2017. Follow Dr. Mirza on twitter @kmirza.