Case history
The patient is a 69-year-old man with a history of high-risk MDS (MDS-MLD-RS) diagnosed 1 year prior to his current visit. He was successfully treated with chemotherapy and bone marrow transplantation. For the next year, several marrow examinations were normal and chimerism analysis revealed >98% donor cells. Currently, he presents with vague symptoms and a CBC demonstrates marked thrombocytopenia of 4K/μL. The low platelet count is initially thought to be related to GVHD; however, a bone marrow examination is performed to assess the status of his disease.

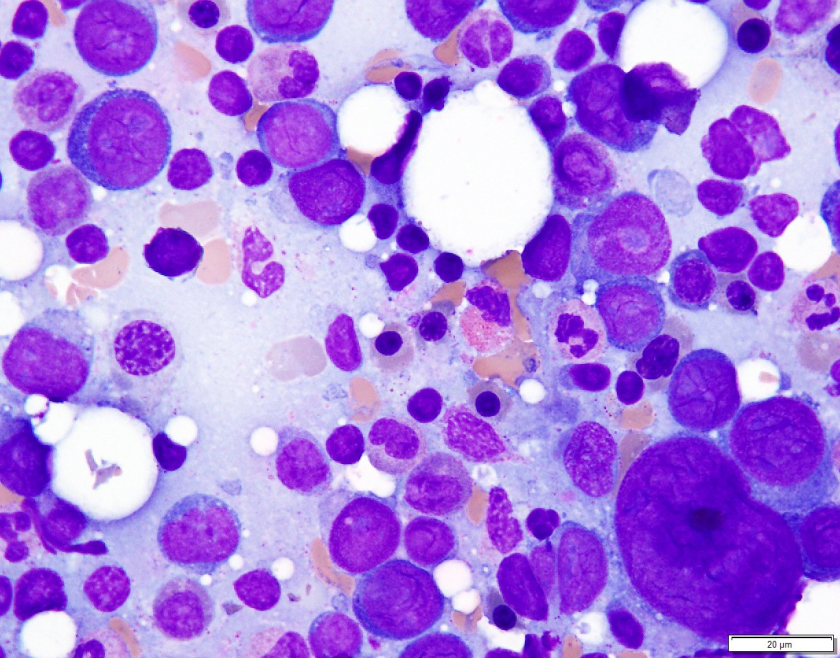

Microscopic Description
Examination of the bone marrow reveals a markedly hypercellular marrow for age with a proliferation of abnormal erythroid cells comprised of sheets of immature and maturing red cell precursors with basophilic cytoplasm. There is a marked increase in larger cells with deeply basophilic cytoplasm, prominent nucleoli, dispersed chromatin, perinuclear hoffs, and a high nuclear to cytoplasmic ratio consistent with pronormoblasts. These pronormoblasts comprised 31% of a 500-cell cell count. Additionally, the background marrow revealed a total of 81% erythroid precursors with marked morphologic atypia and dyspoiesis. Significant dysmegakaryopoiesis is noted. There is no significant increase in myeloid blasts.
Immunophenotyping
Immunohistochemical staining for E-cadherin, CD61 and CD34 is performed. These stains confirm no increase in CD34 positive blasts. CD61 highlights numerous dyspoietic megakaryocytes with widely separated nuclear lobes. E-cadherin staining is impressive, with over 80% of marrow cellularity shown to be comprised of E-cadherin positive erythroid cells.
Diagnosis
The patient’s history of MDS with current dyspoiesis, presence of >80% immature erythroid precursors with >30% proerythroblasts is diagnostic of Acute Myeloid Leukemia, NOS (Pure Erythroid Leukemia) per 2017 revision of the World Health Organization classification of myeloid neoplasms.
While successive chimerism reports thus far had shown >98% donor cells, the chimerism associated with this marrow biopsy reveals a decrease in the percentage of donor cells to 44% confirming the relapsed nature of his myeloid malignancy.
Discussion
Di Guglielmo syndrome, known as M6 leukemia in the FAB classification, was named after Giovanni Di Guglielmo, an Italian hematologist who first characterized the disease in 1917. After a few iterations in different classification schemes, the 2008 WHO Classification characterized two types of ‘erythroleukemia’ the erythroid/myeloid type and the pure erythroid leukemia. The former category of erythroid/myeloid type was removed in the 2017 update of the WHO classification with cases meeting criteria for that diagnosis now falling under the category of MDS. ‘Pure Erythroid Leukemia’ remains, and comes under the AML, NOS category, requiring >80% erythroid progenitors with > 30% proerythroblasts.
An extremely rare leukemia, PEL usually occurs as a progression of previous MDS and very uncommonly as de novo disease. Morphologically, PEL reveals proerythroblasts with deeply basophilic, agranular cytoplasm which is usually vacuolated. Occasionally, smaller ‘blasts’ with scant cytoplasm may resemble lymphoblasts. PEL is an exception to the rule of needing 20% ‘myeloid blasts’ to make an acute leukemia, since often the true myeloblast count is low.
In trephine core biopsies erythroid progenitors may take up an intra sinusoidal growth pattern with a sheet-like arrangement and typically reveal some element of background dysmegakaryocytopoiesis. When PEL lacks specific erythroid differentiation, it may be difficult to differentiate from other types of AML such as Acute Megakaryoblastic Leukemia. Park and colleagues recently categorized some under reported morphologic features of PEL and recurrent cytogenetic abnormalities associated with this disease. These findings included (but were not limited to) a broad morphologic spectrum of erythroblast morphology from undifferentiated blasts to proerythroblasts. They reported bone marrow tumour necrosis in trephine biopsies in over 70% of their cases. Of the cases wherein karyotyping was available, there was a highly complex and monosomal karyotype noted involving the TP53 gene locus.
PEL is associated with an aggressive course with a median survival of 3 months.
References
- Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, Bloomfield CD, Cazzola M, Vardiman JW. The 2016 revision to the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia. Blood. 2016 Jan 1:blood-2016.
- Wang W, Wang SA, Jeffrey Medeiros L, Khoury JD. Pure erythroid leukemia. American journal of hematology. 2017 Mar 1;92(3):292-6.
- Park DC, Ozkaya N, Lovitch SB. Acute leukaemia with a pure erythroid phenotype: under-recognized morphological and cytogenetic signatures associated universally with primary refractory disease and a dismal clinical outcome. Histopathology. 2017 Aug;71(2):316-321. doi: 10.1111/his.13207. Epub 2017 May 5.

-Michael Moravek, MD is a 2nd year anatomic and clinical pathology resident at Loyola University Medical Center. Follow Dr. Moravek on twitter @MoravekMD.

-Kamran M. Mirza, MD PhD is an Assistant Professor of Pathology and Medical Director of Molecular Pathology at Loyola University Medical Center. He was a top 5 honoree in ASCP’s Forty Under 40 2017. Follow Dr. Mirza on twitter @kmirza.
