Case History
16 year old male with a history of chronic pilonidal cyst presented with fatigue, fevers and weight loss. He was febrile and noted to have cervical and inguinal adenopathy. Labs were significant for a white count of 77,000 with 85% peripheral blasts, anemia and thrombocytopenia.
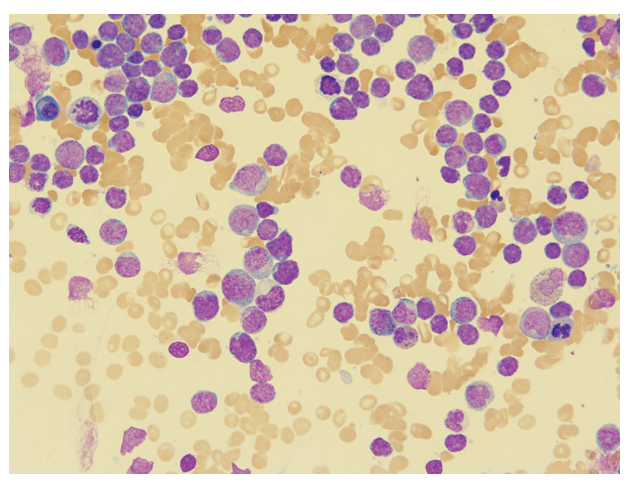




Diagnosis
The bone marrow aspirate shows cellular spicules with sheets of intermediate-to-large sized mononuclear cells with irregular nuclei, distinct nucleoli, dispersed chromatin, and scant to generous amphophilic cytoplasm, with occasional vacuoles, consistent with blasts.
The bone marrow core biopsy shows a greater than 95% cellular marrow, hypercellular for age with approximately 90% of the cellularity composed of an interstitial population of intermediate-to-large sized mononuclear cells with irregular nuclei, distinct nucleoli, dispersed chromatin, and scant to generous amphophilic cytoplasm, with occasional vacuoles, consistent with blasts.
Flow cytometry shows leukemic cells that express immaturity markers (TdT, CD34, CD117, HLA-DR), T cell lineage markers (CD2, CD7 cCD3), and multiple myeloid markers (CD13, CD117, and variable CD15 and CD11b as well as MPO in a small subset).
Bone marrow core biopsy staining (not shown) had similar findings with blasts showing dim-to-strong positivity for myeloperoxidase, lysozyme, CD34 and CD117, as well as strong positivity for TdT. CD7 was weakly positivity, as well as CD3. CD4 and CD5 were negative.


With the expression of MPO by flow cytometric analysis and immunohistochemistry, a final diagnosis of acute leukemia with myeloid and T lymphoid phenotypic features, most consistent with T/Myeloid Mixed Phenotype Acute Leukemia (MPAL) was rendered.
Discussion
Most acute leukemias are definitively assigned to either myeloid, T or B lymphoid lineages. However, approximately 2-5% of patients diagnosed with acute leukemia display an ambiguous lineage after immunophenotyping. A portion of these cases are classified under the category of mixed phenotype acute leukemia (MPAL) by the current WHO nomenclature.1
In a study of 117 MPAL patients by Yan et al, 55% of the cases had combined B/Myeloid, while 33% had T/Myeloid, and 12% had B/T/Myeloid. CD34 was strongly positive in 82% of cases, which reinforces the idea that the cell of origin is a multi-potent stem cell capable of differentiating into both myeloid and lymphoid progenitors. Cytogenetic analysis revealed no chromosomal abnormality in 36% of the patients with MPAL, while 64% had complex karyotypes (>3 aberrations). Translocation (9;22) was the most common abnormality, found in 15% of patients. Monosomy 7, a common finding in myelodysplastic syndromes as well, was found in 7.6% of patients. Mutational analysis revealed IKZF1 deletions in 13% of patients, ASXL1 in 6.5% of patients and a variety of other mutations including ETV6, NOTCH1 and TET2.2
In 2016, Eckstein and colleagues demonstrated epigenetic regulatory genes such as DNMT3A, IDH2, TET3 and EZH2 are the most commonly mutated in MPAL. RAS mutations including NRAS and KRAS and tumor suppressors, such as TP53 and WT1, were frequently identified as well.3
Interestingly enough, the genetic features of MPAL often overlap with early T-cell precursor acute lymphoblastic leukemia (ETP-ALL). ETP-ALL is a high-risk subgroup, representing 10% of adult T-lineage acute lymphoblastic leukemia. It is defined by a characteristic immunophenotype (CD1a/CD8 negative with weak CD5) and distinct gene expression associated with early arrest in T-cell development. This subgroup, called the LYL1 group, expresses the early hematopoietic marker CD34 as well as myeloid antigens (CD13 or CD33), but lacks expression of both CD4 and CD8. These leukemias are associated with a poor prognosis, with a 10- year overall survival of 19% compared to 84% for all other T-ALLs.4
Zhang et al in 2012 performed whole genome sequencing on ETP-ALL cases and found a high frequency of mutations in factors mediating cytokine receptor, tyrosine kinase and RAS signaling. It also showed inactivating mutations in genes encoding transcription factors (GATA3, ETV6, RUNX1, IKZF1) as well as genes involved in histone modification, such as EZH2.5
Overall, the genetic features of both ETP-ALL and MPAL display an identical genomic pattern that involves multiple pathways, including tyrosine kinase signaling, cytokine receptor response, RAS pathway activation, and loss of function in tumor suppressors. These findings give credence to the hypothesis that the early T-cell precursor actually displays more of a pluripotent stem cell profile that is similar to myeloid neoplasms, thus confounding findings found during molecular profiling. With this paradigm in mind, molecular diagnostics cannot differentiate between ETP-ALL and in this case, MPAL.
References
- Swerdlow, Steven H. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Revised 4th ed., International Agency for Research on Cancer, 2017.
- Yan et al. Clinical, immunophenotypic, cytogenetic, and molecular genetic features in 117 patients with mixed-phenotype acute leukemia defined by WHO-2008 classification. 2012 November;97(11):1708-12.
- Eckstein OS et al. Mixed Phenotype Acute Leukemia (MPAL) Exhibits Frequent Mutations in DNMT3A and Activated Signaling Genes. Exp Hematol. 2016 August; 44(8):740-744.
- Ferrando AA et al. Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell. 2002. 1:75–87.
- Zhang J et al. The genetic basis of early T-cell precursor acute lymphoblastic leukemia. Nature. 2012 Jan 11;481(7380):157-63.

–Chelsea Marcus, MD is a third year resident in anatomic and clinical pathology at Beth Israel Deaconess Medical Center in Boston, MA and will be starting her fellowship in Hematopathology at BIDMC in July. She has a particular interest in High-grade B-Cell lymphomas and the genetic alterations of these lymphomas.

Nice case with awesome presentation case thank you alot
This interesting case thank you for sharing …
keep giving👍
Nice case with good presentation I love this kind of cases
Keep giving