This last month, I rotated through our Children’s hospital,
which included reviewing hemoglobin electrophoresis tests. I’d learned about
them before in residency, but they can be quite more interesting (complicated)
than I expected.
Hemoglobin electrophoresis is a blood test to look at
different types of hemoglobin to determine if there are any abnormalities. In a
children’s hospital it is frequently ordered as a reflex for an abnormal
newborn screen or when a child is incidentally found to be anemic. The test is
performed in 2 stages. 1st lysed blood samples are run on gel
electrophoresis and different types of hemoglobin are separated as they move at
different speeds. Several types of hemoglobin will run within the same region,
so a secondary method of separation is always employed.
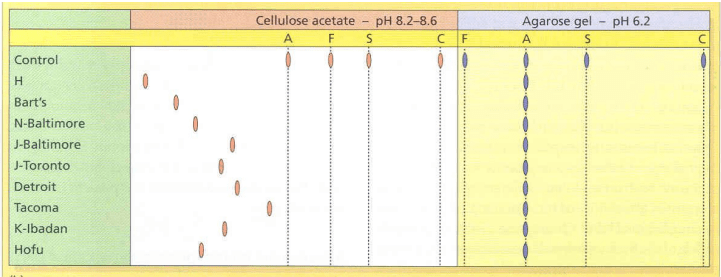
Below, you can see how some bands in the same area of an acidic gel (agarose) are actually very
different on the alkaline gel (cellulose
acetate) and vice versa.
At our hospital, we use HPLC
and measure retention times of the hemolysate to quantify and identify
different hemoglobin types present. As a basic primer you should recall that hemoglobin is a tetramer with a pair of alpha globin + a pair of either beta, delta or gamma globin
(each separate genes).
Alternative hemoglobins are enriched in populations where
malaria is endemic as these variants may provide improved fitness by promoting
resistance to the malarial parasite that reproduces inside red blood cells. Thus,
many people of African or south east Asian descent may carry these variants.
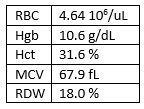
Our case is that of a 2 year old girl with anemia who had
testing sent by her primary care doctor for the following CBC:
This is indicative of microcytic anemia, but unlike some Thalessemias
the RBC isn’t very high. More on this later.
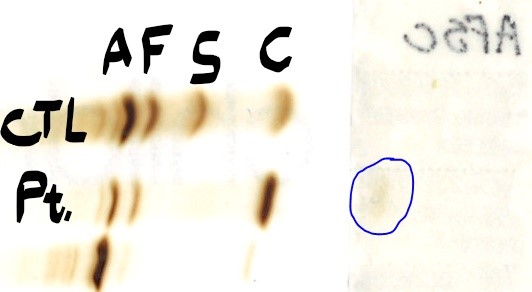
Looking at the gel result, there is a large band in the area
coinciding with Hgb C. We also see the normal Hgb A2 and a small amount of Hgb
F. We know Hgb F can be increased in Hgb SS and thus could also be present if
she had Hgb C trait or disease.
Looking at the next HPLC result, we see there is a similar very high level of Hgb C (68%) with corresponding levels of Hgb F and Hgb A2 (note: acetylated Hgb F and Hgb F are added together). Thus, this fits with a homozygous C with some compensatory A1 and F, right?
Remember Hgb C is a β -globin variant and
you only have 2 β -globin genes, so if you are homozygous
for the C variant on the β-globin gene (HBB), then Hgb A1, which is
made of normal β-globin would be impossible to produce.
Also you might be bothered by all of these small peaks. However, there are
often small peaks that can’t be definitively identified and are likely
post-translationally modified hemoglobin. But in the context of an abnormal Hgb
A1 that shouldn’t be there, we dug deeper.
One of the most common hemoglobinopathies is Beta Thalassemia (β-Thal), which clinically manifests when less of
the beta hemoglobin protein is produced. Heterozygous mutations lead to Beta Thalassemia
minor with minimal symptoms, while homozygous mutations lead to β-thal major with symptoms of anemia. Mutations in the β -globin gene, HBB,
can lead to complete loss of β-globin (β0 variant)
or partial of β-globin (β+ variant).
As this patient has less than 50% of Hgb A present (expected
amount), they could also have a β+ variant
as well. This would make them compound heterozygous for C and β+.
One of the hallmarks of Thalassemia is an increase in Hgb A2 (normal 2.5-3.5%).
Hemoglobin A2 is a normal variant of A that is composed of two alpha and two
delta chains (δ2α2). We see in our
case that the Hgb A2 is normal at 2.5%. So it seems the patient doesn’t display
a typical Thalassemia picture.
One condition that could create this scenario is if there is
a variant in the delta chain of A2
that causes it to elute differently. Indeed, there is a delta variant that
creates hemoglobin A2 prime (A2’)
that moves near the S region of the
HPLC. And when we look back at our unknown hemoglobins, Hgb X is marked at 1.03 of the S region and has an abundance of
3.9%. This supports it being the Hgb A2’ and if we add this together with the
Hgb A2 we get an elevated 6.6% A2 total,
which would be consistent with Beta Thalassemia.
Lastly, one would wonder if we could find this
third hemoglobin variant A2’ on the alkaline gel. Previous studies have shown
the A2’ variant is more negatively charged, so on a basic gel, it should move
further from the negative anode than the other hemoglobins. We don’t see
anything to the left of the HgbC, but if we flip the gel over and look under the patient label, you can see a
faint band that is likely the A2’!
In summary this case arose from 3 separate mutations in a
single patient. She was compound
heterozygous for a Hgb C and β+ variants in the β-globingene and she was heterozygous
for an A2’ variant on the delta-globin
gene. This was certainly a case
where paying close attention mattered.
References:
Abdel-Gadir D, Phelan L, and Bain BJ.
Haemoglobin A2′ and its significance in beta thalassaemia diagnosis. Int J Lab
Hematol. 2009 Jun;31(3):315-9. doi: 10.1111/j.1751-553X.2008.01038.x. Epub 2008
Feb 21.
-Dr. Charles Timmons MD PhD is a pediatric pathologist at Children’s Medical Center in Dallas, TX. His responsibilities include signing out hemoglobin electrophoresis, HPLC and globin sequencing, and has been residency director for 17 years.
-Jeff SoRelle, MD is a Chief Resident of Pathology at the
University of Texas Southwestern Medical Center in Dallas, TX. His
clinical research interests include understanding how the lab intersects
with transgender healthcare and improving genetic variant
interpretation.
An 80 year old man presented with rapid onset of cervical adenopathy over a period of few months. The largest lymph node measuring 6 cm was biopsied and sent for histopathological evaluation.
Biopsy Findings
Sections
from the lymph node showed effacement of the lymph node architecture by a
fairly monotonous population of medium to large sized lymphoid cells arranged
in vague nodular pattern. Focally, a starry sky pattern was observed. The cells
were 1.5-2 times the size of an RBC, with high N:C ratio, irregular angulated
nuclei and small nucleoli. A high mitotic rate of 2-3 mitoses/hpf was seen.
Immunohistochemistry
Immunohistochemical
stains showed that the lymphoma cells were positive for CD20, CD5, SOX-11, and
negative for Cyclin D1, CD10, CD23, CD30, BCL-1, and BCL-6. Ki67 index was
about 70%.
Diagnosis
A diagnosis
of Mantle cell lymphoma, pleomorphic variant was made.
Discussion
Mantle cell
lymphoma is a peripheral B cell lymphoma, occurring in middle aged or older
adults, with a male: female ratio of 7:1. Although Cyclin D1 expression is
considered a hallmark of mantle cell lymphoma, yet about 7% cases are known to
be Cyclin D1 negative. In these cases, morphological features and SOX-11
positivity helps in establishing a definitive diagnosis.
Differential
Diagnosis
In the
assessment of morphological features of lymphoma, the cell size is an important
starting point. In this case, the lymphoma cells ranged from medium to large
sized. The following differential diagnoses were considered:
Burkitt lymphoma
This case showed a “starry sky” pattern focally. A medium sized
population of cells, high mitotic rate and a high Ki67 index (70%) favoured a
Burkitt lymphoma. However, although commonly seen in Burkitt lymphoma, a
“starry sky” pattern is not specific for this type of lymphoma. Also, the lack
of typical “squaring off” of nuclei, basophilic cytoplasmic rim were against
the diagnosis of Burkitt lymphoma. The nuclei in this case showed 0-1 small
nucleoli, unlike the typical basophilic 2-3 prominent nucleoli of Burkitt
lymphoma. Moreover, Ki67 index, even though high was not enough for Burkitt
lymphoma where it approaches 100%. The cells were negative for CD10 and Bcl-6, which
are almost always found in a Burkitt lymphoma. Hence, a diagnosis of Burkitt
lymphoma was ruled out.
Diffuse Large B cell Lymphoma
The presence of interspersed large cells with nucleoli, irregular
nuclei, high mitotic rate, and a high Ki67 index with a history of very rapid
enlargement of lymph node suggested a diagnosis of Diffuse Large B cell
lymphoma. However, the scant cytoplasm, lack of bizarre cells, and absence of
CD10, BCl-2, BCl-6 were against a diagnosis of DLBCL.
Lymphoblastic lymphoma
A diagnosis of lymphoblastic lymphoma was favoured by the irregularly
angulated nuclei, and presence of nucleoli. However, the cells of lymphoblastic
lymphoma have a more delicate nuclear chromatin, higher mitotic rate as against
the relatively condensed chromatin and the low to high variable mitotic rate of
Mantle cell lymphoma. Also, lymphoblastic lymphomas are more commonly of the T
cell subtype and occur commonly in younger individuals. In this case, B cell
markers were positive (CD 20), and the patient was 80 year old, disfavouring a
lymphoblastic lymphoma. The blastoid variant of mantle cell lymphoma is
practically indistinguishable from lymphoblastic lymphoma, except that it is
Tdt negative.
Cyclin
D1 negativity in Mantle cell lymphoma
In the
cases of Cyclin D1 negative mantle cell lymphomas, morphology plays a critical
role in coming to a diagnosis of mantle cell lymphomas. In this case, points
that favoured the diagnosis of mantle cell lymphoma were clinical features such
as older age (80 years), and male gender, and morphological features such as a
vaguely nodular pattern of growth, irregular nuclei, and 0-1 small nucleoli.
Due to the presence of variably sized cells with distinct nucleoli, a
pleomorphic variant was considered. Even though Cyclin D1 was found to be
negative, the cells were positive for SOX-11.
SOX-11 is a
transcription factor that is not normally expressed in B cells, but is
sensitive and fairly specific for mantle cell lymphomas. It is important to
note that SOX-11 is also positive in 25% Burkitt lymphoma, 100% lymphoblastic
lymphoma, and 66% T-prolymphocytic leukemia. Herein lies the importance of
recognising morphological features, as all of these lymphomas that may express
SOX-11 were ruled on the basis of morphology. A more specific antibody, MRQ-58
may be used for greater specificity. The presence of SOX-11 is considered a
specific biomarker for Cyclin-D1 negative mantle cell lymphomas. In these
cases, there is upregulation of Cyclin D2 or D3 that may substitute for Cyclin
D1 upregulation. But, immunohistochemical detection of Cyclin D2 or D3 is not
helpful for establishing a diagnosis, as other lymphomas are commonly positive
for these markers. Hence, it is important to perform SOX-11
immunohistochemistry to diagnose the Cyclin D1 negative variant of mantle cell
lymphoma.
SOX-11 can
be used not just for the diagnosis, but also for determining prognosis of
mantle cell lymphoma. Indolent MCL usually lack SOX-11 expression. The pattern
of SOX-11 staining has also been used a marker of prognosis. Cytoplasmic
expression of MCl, seen in only a few cases was associated with a shorter
survival as compared to the more common nuclear staining of SOX-11.
Conclusion
In this
age, lymphoma diagnosis relies heavily on the use of immunohistochemical
markers. However, this case highlights the importance of morphological features
in diagnosing lymphomas with unusual immunohistochemical marker profile.
Although, this case was negative for Cyclin D1, considered a hallmark of Mantle
cell lymphoma, yet, the combination of morphological features with SOX-11
staining helped in clinching the diagnosis. To avoid a misdiagnosis, it would
be prudent to perform SOX-11 staining in all lymphoma cases morphologically
resembling MCL, but lacking Cyclin-D1.
-Swati Bhardwaj, MD has a special interest in surgical pathology and hematopathology. Follow her on Twitter at @Bhardwaj_swat.
–Kamran M. Mirza, MD, PhD, MLS(ASCP)CM is an Assistant Professor of Pathology and Laboratory Medicine, Medical Education and Applied Health Sciences at Loyola University Chicago Stritch School of Medicine and Parkinson School for Health Sciences and Public Health. A past top 5 honoree in ASCP’s Forty Under 40, Dr. Mirza was named to The Pathologist’s Power List of 2018 and placed #5 in the #PathPower List 2019. Follow him on twitter @kmirza.
A
4 year old child was brought to the pediatrician by her mother with a complaint
of new onset of severe bruising on her legs. The mother could not recall any
falls or bumps that would have caused the bruising. On exam, the physician also
noted mucosal bleeding in the oral cavity. Questioning revealed that the
patient had experienced flu like symptoms several weeks earlier. The physical
exam was normal except for the bleeding. There was no family history of
bleeding disorders. A CBC was ordered.
Reported
CBC Results
WBC,
RBC, Hgb, Hct, RBC indicies normal
Platelet
count 26 x 103/μL
IPF
22% (reference range IPF% 1.0-7.0%)
The physician evaluated the results, noting the
normal CBC but decreased platelet count. The above results also show the
immature platelet fraction (IPF), an additional Advanced Clinical Parameter reported
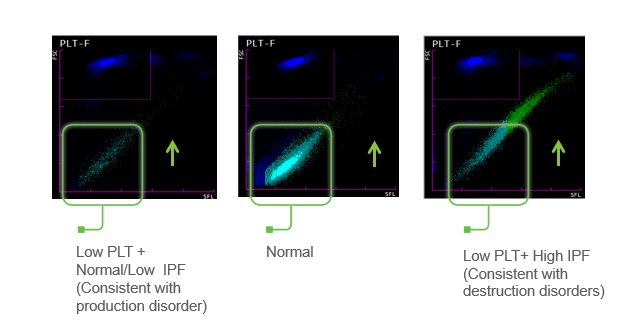
from the Sysmex XN hematology analyzer. A low platelet count, as seen in this
patient, will reflex a fluorescent platelet count (PLT-F). The impedance count (PLT-I)
can be falsely increased if small RBCs or fragments are counted as platelets. On the other hand, in an optical
platelet count, when measuring platelets by size (PLT-O), large platelets can
be missed, giving a falsely low count. In this case there was a low
platelet count and an instrument flag for an abnormal platelet scattergram. The PLT-F, on the other hand, uses a
platelet specific dye which eliminates interference seen with other methods.
The fluorescent dye labels the RNA, and forward scatter is used to determine
size while side fluorescence is used to measure RNA content. With gating set
based on cell volume and RNA content, the PLT-F can be measured. Therefore,
the reflexed and more reliable
PLT-F was the reported count.
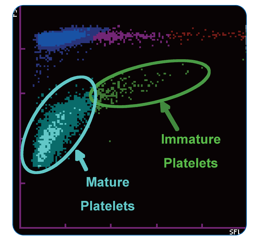
Figure 1. PLT-F scattergram. The PLT-F channel measures forward scatter (FSC) on the Y axis and side fluorescence (SFL) on the X axis.1
Additionally, when there is an abnormal
scattergram or a low platelet count, the IPF% and IPF# are also reported. The immature
platelet fraction is a measure of the youngest platelets, or reticulated
platelets. These are the first circulating platelets, right out of the bone
marrow. An increased IPF indicates an increase in platelet production, yet this
child’s platelet count was very low. This suggests that the thrombocytopenia
may be due to excessive destruction of platelets; the bone marrow was actively
making platelets, but they were being destroyed, causing the low platelet
count.
Figure 2. Platelet scattergrams from a healthy individual with a normal IPF (a) and a patient with a high IPF (b). Mature platelets appear as blue dots, green dots represent the IPF with increased cell volume and higher fluorescence intensity compared to mature platelets.1
Diagnosis
Immune Thrombocytopenia- ITP.
Primary immune thrombocytopenia (ITP), formerly
known as idiopathic thrombocytopenic purpura or immune
thrombocytopenic purpura, is one of the most common bleeding
disorders of children. In most cases, it presents with sudden onset of bruising
and petechiae in an otherwise healthy child, with normal WBC and hemoglobin. ITP is an
autoimmune bleeding disorder in which the immune system makes anti-platelet
antibodies which bind to platelets and cause destruction. Even though the exact
cause of ITP remains unknown, it is recognized that it can follow a viral
infection or live vaccinations. While there are some similarities between
pediatric ITP and ITP in adults, in children this tends to be an acute disease
which is self-limiting and resolves itself in several weeks, with no treatment.
However, in a small number of children, the disorder may progress to a chronic
ITP. In contrast to ITP in children, a chronic form is more commonly seen in
adults. It is usually a diagnosis of exclusion, does not follow a viral illness
and requires treatment.
This
patient recovered in a few weeks. One month after the initial episode, her PLT
was 174 x 103/μL and her IPF% was 6.0%
Conclusion
An IPF reported with a CBC, in combination with a low platelet count, is fast, inexpensive, and can be extremely beneficial in aiding in a timely diagnosis. As the child’s platelet count recovered, the IPF% returned to normal range. ITP can therefore be monitored with a CBC. Thus, the IPF can be used not only to help diagnose but also as an indicator of remission.
Arshi Naz et al. Importance of Immature platelet
Fraction as a predictor of immune thrombocytopenic purpura. Pak J Med Sci 2016
Vol 32 No 3:575-579
Briggs,C. Assessment of an immature plateletfraction
(IPF) in peripheral thrombocytopenia. Br J Haematol 2004Jul;126(1):93-9
Sysmex White Paper. The role of the ImmaturePlatelet
Fraction(IPF) in the differential diagnosis of thrombocytopenia. www.sysmex.com/us
D-Orazio, JA, Neely, J, Farhoudi,N. ITP in children: pathophysiology and current
treatment approaches.J Pediatr Hematol Oncol.2013 Jan;35(1): 1-13
-Becky Socha, MS, MLS(ASCP)CM BB CM graduated from Merrimack College in N. Andover, Massachusetts with a BS in Medical Technology and completed her MS in Clinical Laboratory Sciences at the University of Massachusetts, Lowell. She has worked as a Medical Technologist for over 30 years. She’s worked in all areas of the clinical laboratory, but has a special interest in Hematology and Blood Banking. When she’s not busy being a mad scientist, she can be found outside riding her bicycle.
The patient is a 69 year old male who presented to the
hospital with a 3-month history of drenching night sweats, weight loss, fatigue,
and generalized lymphadenopathy. He also endorsed a very itchy rash all over
his body. He denied smoking. There was no other relevant social or family
history.
Physical examination confirmed diffuse lymphadenopathy,
hepatosplenomegaly and a mild diffuse skin rash. Notably, there was a 2.5 cm
level-1 lymph node palpated in the left neck. This was subsequently biopsied.
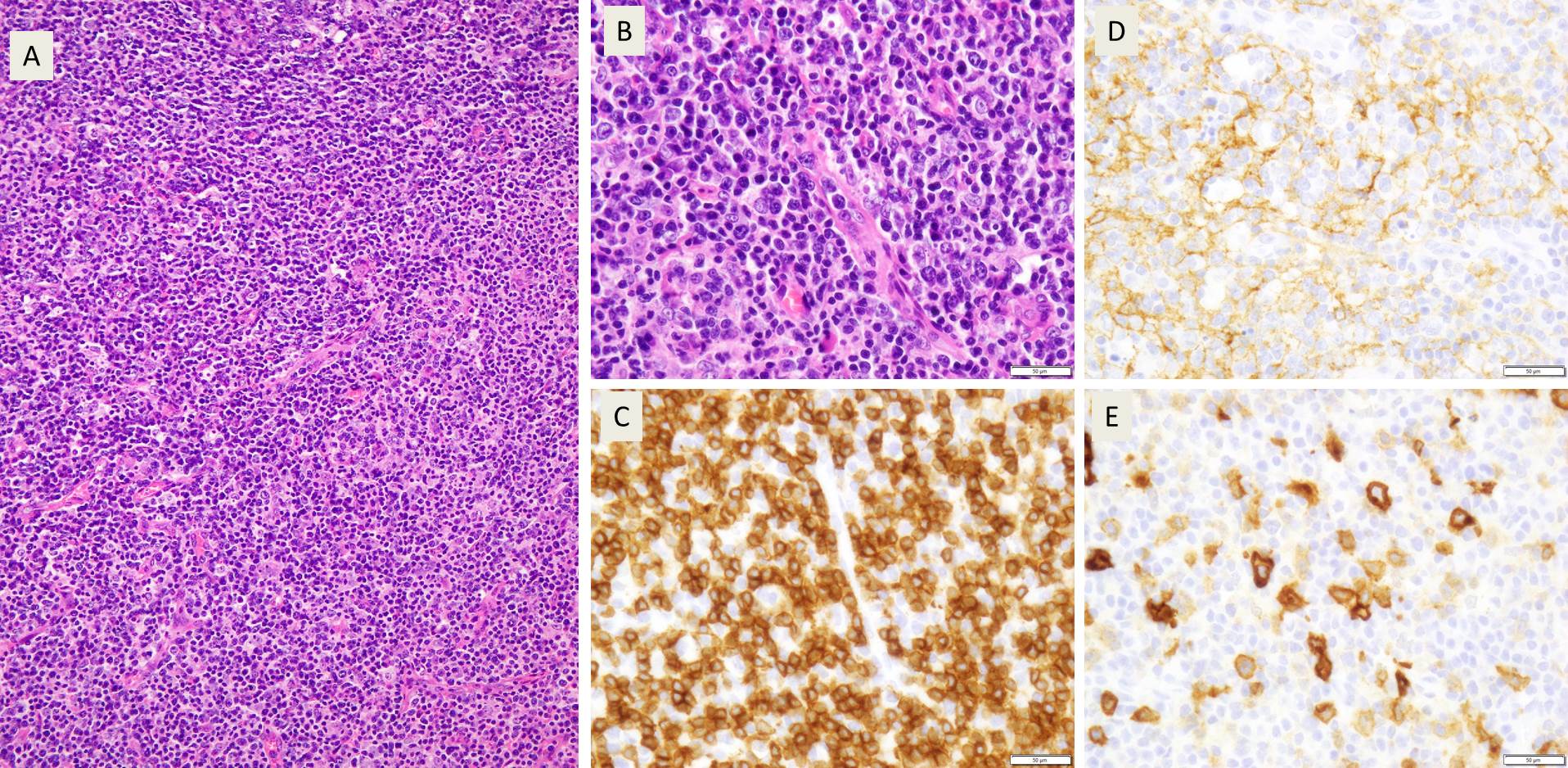
Biopsy
Biopsy of the level-1 neck lymph node revealed a 2.3 x 1.5 x
1.2 cm mass pink-tan and firm mass. Sectioning revealed a glossy white-tan cut surface.
H&E staining revealed a polymorphic lymphocytic infiltrate of in the interfollicular
zones. The infiltrating lymphocytes ranged from small to large cells with abundant
cytoplasm, eosinophils, and plasma cells. There was also a notable increase in
the number of high endothelial vessels lined by lymphocytes with irregular
nuclear borders and clear cytoplasmic zones.
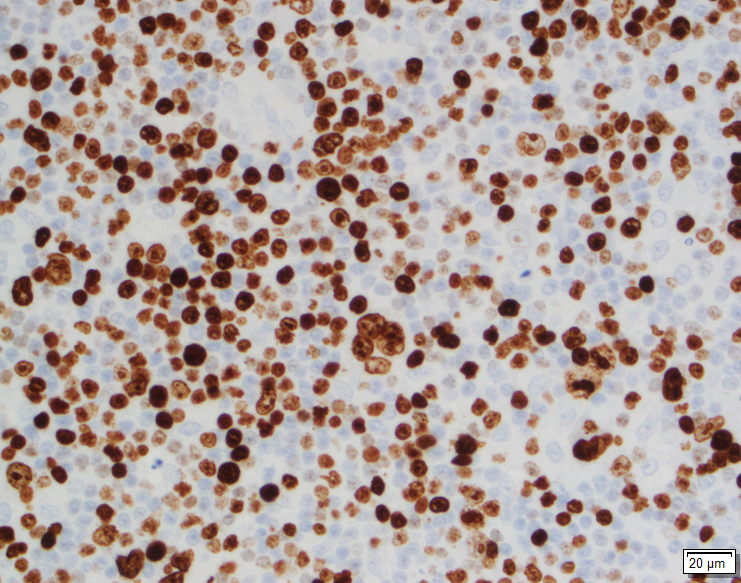
Image 1. Polymorphic infiltrate of small, mature appearing lymphocytes (A), with prominent blood vessels and clear cytoplasm (B). Most of these cells were CD3 positive T cells (C) with expanded CD21 positive FDC meshworks (D) and scattered CD30 positive immunoblasts (E)
Further characterization by immunohistochemical staining showed
the majority of the interfollicular cells to be CD3 and CD5 expressing T cells.
These were a mix of CD4 and CD8 positive cells but with marked CD4
predominance. CD7 appeared positive in a smaller population of T-cells compared
to CD3 (consistent with loss of this pan-T-cell marker). Varying numbers of the
interfollicular cells were positive for CD10, BCL-6, CXCL-13, and PD-1 with a
strong positivity for ICOS, phenotypically consistent with an expansion of Tfh
(T-follicular helper cell) cells.
Interspersed between the T cells were numerous CD20 positive
cells with prominent nucleoli that also revealed CD30 positivity. CD21 staining
revealed expanded follicular dendritic cell meshworks. EBER ISH was positive in
a rare subset of cells. Kappa and lambda ISH showed an increased number of
polytypic plasma cells.
Flow Cytometry showed the presence of a small population of
T-cells that were CD4 positive but CD3 negative. There was no evidence of
B-cell clonality. TCR-G PCR was positive.
A final diagnosis of Angioimmunoblastic T-cell lymphoma
(AITL) was rendered.
Discussion
AITL is a relatively rare neoplasm of mature T follicular
helper cells, representing about 1-2% of all non-Hodgkin lymphomas. It is;
however, one of the more common subtypes of peripheral T-cell lymphomas,
accounting for 15-30% of this subgroup. The condition was first reported in
1974 in Lancet as a non-neoplastic abnormal immune reaction1. It was
first recognized as a distinct clinical entity in in 1994 in the Revised
European American Lymphoma Classification2. The disease shows a
geological preference to Europe (28.7%) over Asia (17.9%) and North America
(16%). AITL occurs primarily in middle aged and elderly individuals and shows a
slight predominance of males over females.
The disease has a strong association with EBV infection, but
the neoplastic T-cells are almost always EBV negative, creating an interesting
question of EBV’s function in the etiology of AITL. AITL most often presents
late in the disease course with diffuse systemic involvement, including
hepatosplenomegaly, lymphadenopathy and other symptoms such as rash with
pruritis and arthritis. Lab findings include cold agglutinins, rheumatoid
factor and anti-smooth muscle antibodies. There also tends to be
immunodeficiency secondary to the neoplastic process. The clinical course of
AITL is variable, but the prognosis is poor, with the average survival time
after diagnosis being < 3 years. The histological features and genetic
findings have not been found to impact clinical course.
Microscopically, AITL presents with either partial or total
effacement of the normal lymph node architecture with perinodal infiltration.
The cells of AITL are small to medium-sized lymphocytes with clear to pale
cytoplasm, distinct cell membranes and very minimal cytological atypia. These
cells often congregate around the high endothelial venules. The T-lymphocytes
are present in a largely polymorphous inflammatory background of other
lymphocytes, histiocytes, plasma cells and eosinophils. There are 3 overlapping
sub-patterns of AITL. The first of these is similar to a reactive follicular
hyperplasia, and can only be distinguished from normal hyperplasia by use of
immunohistochemical stains to differentiate the neoplastic cells from normal
reactive cells. The second pattern has retained follicles, but they show
regressive changes. The third pattern has completely or sub totally effaced.
These three patterns seem to be on a spectrum with one another, given that
progression from the first to the third pattern has been seen on consecutive
biopsies in the same patient.
Cytologically, AITL cells express pan-T-cell markers
including CD2, CD3 and CD5 and the vast majority are CD4 positive. CD3 may be
quantitatively decreased or absent by flow cytometry. There are a variable
number of CD8 positive T-cells. The tumor cells also show the immunophenotyping
of normal T follicular helper cells including CD10, CXCL13, ICOS, BCL6 and PD1
in 60-100% of cases. CXCL13 and CD10 are the most specific, whereas PD1 and
ICOS are the most sensitive.
References
Horne, C., Fraser, R., & Petrie, J. (1974).
Angio-Immunoblastic Lymphadenopathy With Dysproteinemia. The
Lancet, 304(7875), 291. doi:10.1016/s0140-6736(74)91455-x
Harris, N.l. “A Revised European-American
Classification of Lymphoid Neoplasms: a Proposal from the International
Lymphoma Study Group.” Current Diagnostic Pathology, vol. 2, no. 1, 1994,
pp. 58–59., doi:10.1016/s0968-6053(00)80051-4.
Swerdlow, Steven H. WHO Classification of
Tumours of Haematopoietic and Lymphoid Tissues. International Agency for
Research on Cancer, 2017.
-Zachary Fattal is a 4th year medical student at the Central Michigan University College of Medicine. He is pursuing a career in pathology and has a special interest in hematopathology, cytopathology and blood bank/transfusion medicine. You can follow him on Twitter @Paraparacelsus.
–Kamran M. Mirza, MD, PhD, MLS(ASCP)CM is an Assistant Professor
of Pathology and Medical Education at Loyola University Health System. A
past top 5 honoree in ASCP’s Forty Under 40, Dr. Mirza was named to
The Pathologist’s Power List of 2018. Follow him on twitter @kmirza.
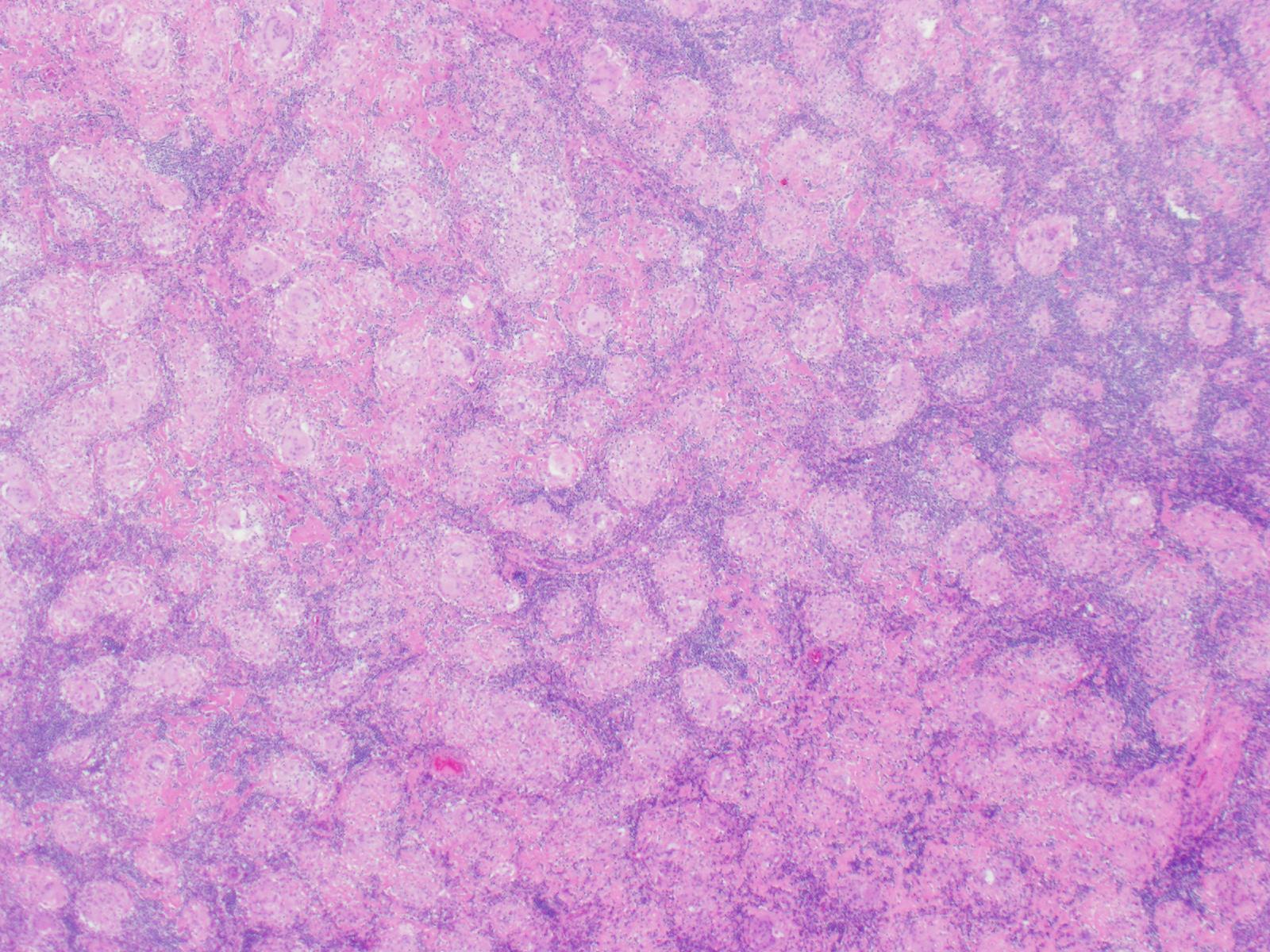
A 36 year old female underwent thyroidectomy for multinodular goitre that led to the fortuitous discovery of a neck mass. The neck mass specimen submitted comprised two lymph nodes measuring 2.2 cm and 1.3 cm in the greatest dimensions, with a fleshy tan cut surface.
Biopsy Findings
H&E stained sections revealed numerous non-necrotizing
granulomas effacing and replacing normal lymph node architecture. These
consisted of pale epithelioid histiocytes and Langhans type of giant cells. The
granulomas lacked a peripheral rim of lymphocytes. AFB and GMS stains were
negative for microorganisms
Diagnosis
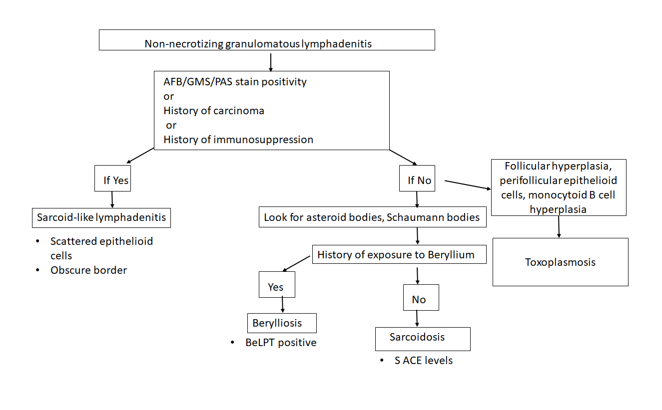
A diagnosis of non-necrotizing granulomatous
lymphadenitis was rendered noting that in the correct clinical context the
findings could represent sarcoidosis.
Discussion
Granulomatous inflammation is a special type of
chronic inflammatory response characterised by the formation of discrete
collections of histiocytes called granulomas. Activated histiocytes appear as
epithelioid cells with round to oval nuclei, often with irregular contours and
abundant granular eosinophilic cytoplasm with indistinct cell borders. They may
coalesce to form multinucleated giant cells. When found in the lymph node, the
reaction pattern is called granulomatous lymphadenitis. It can be caused by a
variety of different conditions, and therefore, requires thorough workup to
come to a conclusive diagnosis.
On the basis of presence or absence of necrosis,
granulomatous lymphadenitis can be classified as necrotizing or non-necrotizing.
Additionally, the presence of an abscess, usually central, indicates a
suppurative lymphadenitis.
Non-necrotizing
granulomatous lymphadenitis:
Sarcoidosis lymphadenitis is
the prototype of non-necrotizing granulomatous lymphadenitis. It shows the
presence of discrete granulomas without a peripheral rim of lymphocytes, called
“naked granulomas”. The early phase shows follicular hyperplasia and sinus
histiocytosis, followed by appearance of epithelioid cell nodules toward the
end of this phase. The peak phase shows well-demarcated
granulomas composed of epithelioid cells with scattered multinucleated giant
cells observed throughout the lymph node. Granulomas may occasionally coalesce.
In the late phase, increased collagen fibers result in fibrosis and
hyalinization. There are no neutrophils and it is uncommon to find small
foci of central necrosis. Numerous inclusions such as asteroid, Schaumann, or
Hamazaki-Wesenberg bodies can be seen. In this case, we observed
well-demarcated granulomas throughout the lymph node, typical of the peak phase
without any caseous necrosis or suppuration.
Other causes of granulomatous lymphadenitis can be ruled
out as follows.
Sarcoid-like lymphadenitis:
It shows a similar pattern of non-necrotizing
lymphadenitis like sarcoidosis. However, classically sarcoid like reaction
shows scattered small epithelioid granulomas with sparsely arranged epithelioid
cells. The border of the granulomas is usually obscure. The CD4:CD8 ratio
ranges from 0.8 to 2.25 while in sarcoidosis, it is >3.5. These findings
help distinguish sarcoid-like lymphadenitis from sarcoidosis.
Sarcoid-like adenitis may be seen in numerous conditions
such as carcinoma, Toxoplasmosis, fungal infections, tuberculosis,
immunocompromised states, pneumoconiosis etc. The fact that tuberculosis and
fungal infections can present with a non-necrotizing granulomatous lymphadenitis
highlights the importance of performing fungal (PAS & GMS) and AFB (Ziehl
Neelson) stains in non-necrotizing lymphadenitis as well. In this case, the
granulomas had distinct borders, numerous epithelioid cells, no organisms were
identified on special stains, nor was there any history of immune compromise;
ruling out a sarcoid-like reaction.
Berylliosis: The lymph node picture in Berylliosis is identical to that of sarcoidosis.
We may even see asteroid bodies or Schaumann bodies. A diagnosis can be
established by eliciting a history of chronic exposure to Beryllium. Beryllium
lymphocyte proliferation test (BeLPT) is a test that measures Beryllium
sensitization and is very specific for Beryllium exposure. There was no known
history of exposure to Beryllium in this case.
Toxoplasmosis: A classic triad of follicular hyperplasia, small granulomas composed of epithelioid cells within and around hyperplastic follicles and, monocytoid B cell hyperplasia, is observed in toxoplasmosis lymphadenitis. This case did not show follicular hyperplasia, ruling out toxoplasmosis.
Necrotizing granulomatous lymphadenitis
Even though we did not find any necrosis in this case,
yet, it is worthwhile to review briefly the various causes of necrotizing lymphadenitis.
Non-suppurative
Tuberculosis: Histology
of a tuberculous lymph node is characterised by central caseous necrosis
surrounded by an epithelioid cell layer. The outermost layer is comprised of
lymphocytes and fibrosis. Plasma cells are not observed. Diagnosis can be
established by performing an AFB stain that demonstrates acid fast rod shaped
bacteria in the areas of necrosis. Organisms can also be detected by PCR.
BCG lymphadenitis:
About 0.7 to 2.3% of BCG vaccinated children may develop BCG lymphadenitis that
is smaller than tuberculous lymphadenitis. Early phase shows follicular
hyperplasia and sinus histiocytosis. Later, there is development of
micronodules of epithelioid granulomas without necrosis and epithelioid cell
granulomas with central caseous necrosis. Langhans giant cells are rare.
Fungal infections:
Fungal infections by Histoplasma, Cryptococcus, coccidiodomycosis, pneumocystis
may also cause a necrotizing granulomatous inflammation. There are numerous
neutrophils, and fungal structures can be seen. GMS and PAS can be used in
cases where it is difficult to the find the fungal elements on H&E.
Suppurative
Tularemia: There
are three forms of histological changes, Abscess form, showing abscess
with central necrosis and mononuclear cells, Abscess-granulomatous form with granulomas with central necrosis,
which form large lesions with central abscesses, and granulomatous form with
caseating necrosis at the centre of the granulomas.
Cat Scratch disease:
Similar to tularemia, there are three phases of histologic presentation, an
early phase of follicular hyperplasia, intermediate phase of microabscess, and
a late phase of granulomatous inflammation. Monocytoid B cell clusters are
observed close to the abscess.
Conclusion
Sarcoidosis is usually diagnosed by
excluding other causes of granulomatous inflammation, as we did in this case.
Characteristic non-necrotizing, discrete granulomas were seen throughout the
lymph node. The age of the patient and female gender epidemiologically support
the diagnosis. This case reflects an example work up of a granulomatous
lymphadenitis that is a morphologic presentation of myriad diseases.
-Swati Bhardwaj, MD has a special interest in surgical pathology and hematopathology. Follow her on Twitter at @Bhardwaj_swat.
–Kamran M. Mirza, MD, PhD, MLS(ASCP)CM is an Assistant Professor of Pathology and Medical Education at Loyola University Health System. A past top 5 honoree in ASCP’s Forty Under 40, Dr. Mirza was named to The Pathologist’s Power List of 2018. Follow him on twitter @kmirza.
A sixty nine year old female who underwent right breast reconstruction about 13 years ago due to breast cancer presents to the doctor office with right breast pain and right breast enlargement over the last two months. She has lost some weight and does not recall any trauma to this area. She had a textured saline implant. Examination reveals no definite palpable masses. MRI of right breast showed intact saline implant with moderate amount of fluid surrounding the implant within the intact external capsule. No adenopathy was noted. Right breast implant was removed and complete capsulectomy was performed.
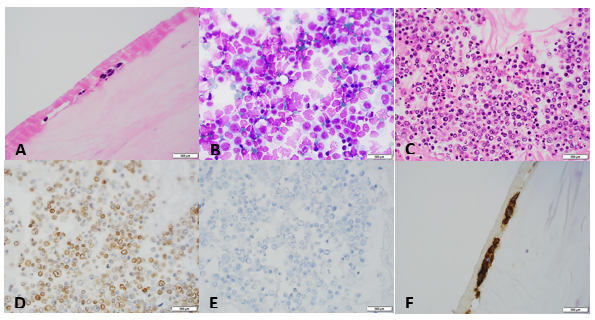
Image 1. A. Section of breast capsule with rare atypical hyperchromatic cells (arrow). B. Cytospin preparation of the fluid surrounding the implant with numerous atypical lymphocytes. C. Cell block of the fluid with large atypical lymphocytes. D, E. Lymphocytes are positive for CD30 (image D) and negative for ALK-1 (image E). F. CD30 positive cells in the section of the implant.
Diagnosis
Breast implant-associated
anaplastic large cell lymphoma.
Discussion
Breast implant associated
anaplastic large cell lymphoma is a provisional entity that is morphologically
and immunophenotypically similar to ALK-negative anaplastic large cell lymphoma.
It arises primarily in association with a breast implant. It is a very rare
entity with an incidence of 1 in 500,000 to 3 million women with implants. Tumor
cells may be localized to the seroma cavity or may involve pericapsular fibrous
tissue. Sometimes it can form a mass lesion. Locoregional lymph node may be
involved. The mean patient age is 50 years. Most patient presents with
stage 1 disease, usually with peri-implant effusion. The mean interval from
implant placement to lymphoma diagnosis is 10.9 years. There is no association
with the type of implant. Histologic examination shows two different types of
proliferations. In patients with seroma, the proliferation is confined to the
fibrous capsule (“in situ” iALCL). However, the distribution of neoplastic
lymphocytes could be heterogeneous with some cellular areas with numerous large
pleomorphic cells of varying size and some fibrotic areas with rare atypical
lymphocytes. It is beneficial to look at the seroma fluid in addition to
capsule sections, because sometimes the neoplastic lymphocytes are
predominantly present in fluid (as in our case). Patients presenting with tumor
mass show more heterogeneous proliferations infiltrating surrounding tissues
(“infiltrative” iALCL). They consists of either sheets are clusters of large
neoplastic cells accompanied by a large number of eosinophils. By immunohistochemistry,
the tumor cells are strongly positive for CD30. CD2 and CD3 are more often
positive than CD5. CD43 is almost always expressed. Most cases are CD4 positive. The
prognosis is very good in patients with disease confined to the capsule.
The median overall survival is 12 years. However, patients with a tumor mass
could have a more aggressive clinical outcome.
References
1. Swerdlow SH, Campo E, Harris NL, et al. WHO Classification of Tumours of
Haematopoetic and Lymphoid Tissues (Revised 4th edition). IARC: Lyon
2017.
2. Jaffe, E , Arber, D, et al. Hematopathology (second edition) 2017.
-Junaid Baqai, MD, was born in Chicago, IL but spent most of his life in Karachi, Pakistan. He graduated from DOW Medical College in Pakistan and did his residency in anatomic and clinical pathology at Danbury Hospital, CT followed by hematopathology fellowship from William Beaumont Hospital, Michigan and oncologic-surgical pathology fellowship from Roswell Park Cancer Institute, New York. He currently serves as Medical Director of hematology, coagulation and flow cytometry at Memorial Medical Center and Medical Director of Laboratory at Taylorville Memorial Hospital.
76 year old man with a history of chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) with new anterior mediastinal mass and increasing lymphadenopathy.
Lymph Node Biopsy
H&E
Diagnosis
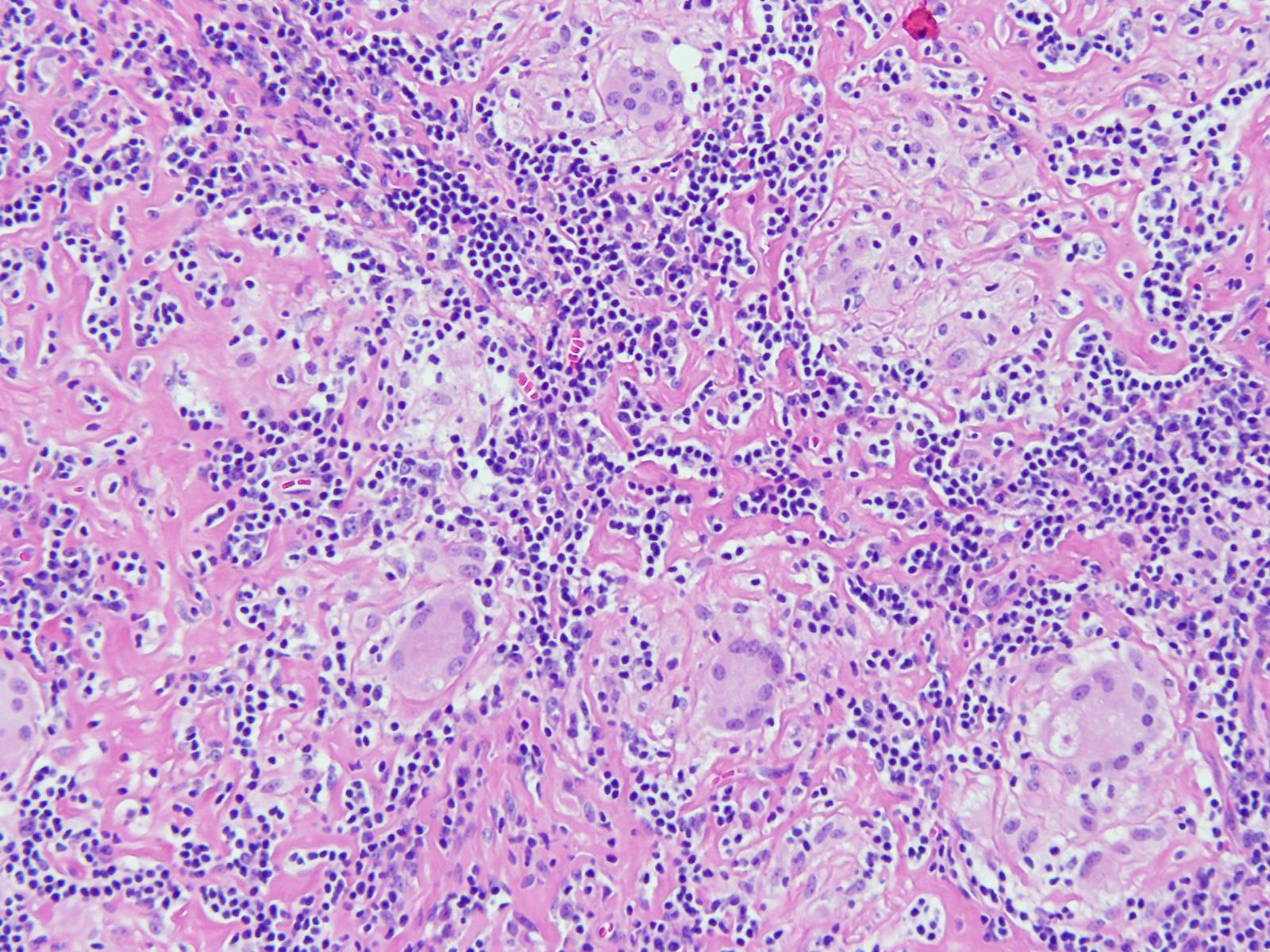
Tissue sections show a diffuse atypical lymphoid infiltrate
that completely effaces the normal nodal architecture. The infiltrate is
composed of numerous small lymphocytes with round to mildly irregular nuclei,
clumped chromatin, inconspicuous nucleoli and scant cytoplasm. There are also
expanded pale areas that contain intermediate sized cells with more open
chromatin and distinct single to multiple nucleoli. These cells are most
consistent with prolymphocytes/paraimmunoblasts and form the proliferation
centers characteristic of CLL/SLL. Occasional centroblastic-type B-cells are
noted within these proliferation centers. In addition, there are scattered single
to multinucleated cells that have irregular nuclear membranes with pale,
vesicular chromatin and prominent inclusion-like, eosinophilic nucleoli. These
cells morphologically resemble Hodgkin cells, Reed-Sternberg cells, mummified
forms and other variants. These large cells are more evident in areas with a
histiocyte rich background and around foci of necrosis. Occasionally, apoptotic
bodies and mitotic figures are seen.
Immunohistochemical studies
show that the vast majority of the small-intermediate lymphocytes express B-cell
markers CD20 (dim) and PAX5 and co-express CD5 and CD23 (subset). This is
consistent with a background of CLL/SLL. The large atypical cells are positive
for CD30, PAX5 and CD20 (variable). CD3 highlights numerous scattered
background small T-cells, which are increased in the areas with the large cells.
In situ hybridization for Epstein Barr viral RNA (EBER ISH) is mainly staining
the large atypical cells. By Ki-67, the proliferation fraction is overall
increased (40%) with increased uptake by the large atypical cells.
The morphologic and immunophenotypic findings are consistent
with involvement by the patient’s known small lymphocytic lymphoma/chronic
lymphocytic leukemia (SLL/CLL) with aggressive morphological features. The
aggressive features include expanded proliferation centers and an elevated
Ki-67 proliferative index (40%). Additionally there are histiocyte/T-cell rich
areas composed of multiple EBV positive large atypical cells with morphologic
and immunophenotypic features compatible with Hodgkin/ Reed-Sternberg cells.
These areas are most in keeping with evolving classic Hodgkin lymphoma. Sheets
of large cells indicative of large cell transformation are not seen, although
increased scattered large centroblastic-type B cells are present.
Discussion
Lymph node involvement by CLL/SLL will typically show a
diffuse proliferation of small lymphocytes with effacement of the normal nodal
architecture. The small lymphocytes have
round nuclei, clumped chromatin and scant cytoplasm. Scattered paler areas
known as proliferation centers are characteristic of this entity. The
proliferation centers are composed of a mixture of cell types including small
lymphocytes, prolymphocytes and paraimmunoblasts. Prolymphocytes are small to
medium in size with relatively clumped chromatin, whereas paraimmunoblasts are
larger cells with round to oval nuclei, dispersed chromatin, eosinophilic
nucleoli and slightly basophilic cytoplasm. Some cases show increased and
enlarged proliferation centers with a higher proliferation rate. This must be
distinguished from large cell transformation.1
Aggressive features of CLL/SLL include proliferation centers
that are broader than a 20x field or becoming confluent. An increased Ki-67
proliferation >40% or >2.4 mitoses in the proliferation centers can also
portend a more aggressive course. These cases tend to have worse outcomes than
typical CLL/SLL and better outcomes than cases that have undergone Richter
transformation to diffuse large B-cell lymphoma (DLBCL). Transformation to
DLBCL occurs in 2-8% of patients with CLL/SLL. Less than 1% of patients with
CLL/SLL develop classic Hodgkin lymphoma (CHL). In order to diagnose CHL in the
setting of CLL/SLL, classic Reed-Sternberg cells need to be found in a
background appropriate for CHL, which includes a mixed inflammatory background.
The majority of these CHL cases will be positive for EBV.1
Richter’s transformation is defined as an aggressive
evolution of CLL. While the most common type of transformation is to a
high-grade B-cell Non-Hodgkin lymphoma, other histological transformations have
been described. This includes CHL, lymphoblastic lymphoma, hairy cell leukemia
and high-grade T-cell lymphomas. The prognosis for patients who present with
transformation to CHL is poor compared to de novo CHL.2
A large study from the M.D. Anderson Cancer Center described 4121 patients with
CLL/SLL and found that only 18 patients or 0.4% developed CHL. The median time
from CLL to CHL diagnosis was 4.6 years. Fourteen of the patients received
chemotherapy. The overall response rate was 44% with a complete response rate
of 19%. The median overall survival was 0.8 years and all patients eventually
died from disease recurrence or progressive disease.3 This dismal
prognosis is similar to patients with Richter transformation to DLBCL and much
worse than patients with de novo CHL, which is curable in >85% of cases.1
References
Swerdlow SH, Campo E, Harris NL, et al. WHO
Classification of Tumours of Haematopoetic and Lymphoid Tissues (Revised 4th
edition). IARC: Lyon 2017.
Janjetovic S, Bernd HW, Bokemeyer C, Fiedler W. Hodgkin’s
lymphoma as a rare variant of Richter’s transformation in chronic lymphocytic
leukemia: A case report and review of the literature. Mol Clin Oncol.
2016;4(3):390–392.doi:10.3892/mco.2016.727.
Tsimberidou, AM, O’Brien, S and Kantarjian, HM,
et. al. Hodgkin transformation of chronic lymphocytic leukemia. Cancer. 2006;107(6).doi.org/10.1002/cncr.22121.
–Chelsea Marcus, MD is a Hematopathology Fellow at Beth Israel
Deaconess Medical Center in Boston, MA. She has a particular interest in
High-grade B-Cell lymphomas and the genetic alterations of these
lymphomas.
Last month, I wrote about some
projects I did while rotating through the pathology program at Danbury
Hospital in Connecticut. This month I’m in a more clinical setting with a
hematology/oncology clerkship at Northwell’s Staten Island University Hospital.
But, over the past few months of rotations (and arguably a lot longer before
medical school) I’ve been noticing a part of laboratory medicine which often
intersects with our clinical colleagues at the bedside. I’ve told you about the
pitfalls and successes in the relationships
between surgeons and anatomic pathologists before, where frozen sections
are critical and time is of the essence. And we’ve all seen collaboration
between the bench and bedside before—think microbiology and infectious disease,
blood bank and literally everyone, etc. Still, one collaborative effort sort of
happens behind the shadows, behind phone calls and lab reports, and sometimes
with no communication at all! So, what kind of vigilante medicine am I talking
about? Who is this Batman of medicine? It’s just our friends in hematology.
When you’re working the hematology bench in the lab, it’s
pretty commonplace for a physician on a hematology service to call and ask for
a peripheral smear to review. Many times, it’s for the purpose of teaching
residents, fellows, or medical students but more often than not it’s a
confirmatory exercise. See, when that hematologist asks to review a slide,
she’s probably coming down to the lab to look at the morphology of red cells
and white cells to help in their differential diagnosis. They might have a
patient with a suspected thalassemia or hemoglobinopathy and, before starting
the full work up of lab tests, just want to see if there are any RBC morphology
traits or target cells that stand out. Thrombocytopenia? Let’s make sure
there’s no platelet clumping. Maybe they’ve got a patient with some kind of
liver or kidney pathology and are on the hunt for acantho- or echinocytes. Or
better yet, someone went hiking, there’s an infectious etiology on their
differential—let’s go hunting for babesia, malaria, oh or even erlichia!
Image 1. Here’s a few examples of three parts of a patient’s smear that are contributory to a particular pathology in vivo. Think you know what it is? I bet you’d be surprised…not all that hyper-segments is a B12/Folate deficiency. But technically it is; read about cobalamin and homocysteine pathology in a neonatal patient here: http://www.bloodjournal.org/content/128/21/2584 (Source: Blood 2016)
I know what you’re thinking. Wait—that’s our job as
medical laboratory scientists; our literal job. Our instruments, that we
validate, and correlate, and make sure work fantastically give us flags. We
investigate those flags and look at smears ourselves! We collaborate with other
lab techs, and with our pathologist colleagues and send out final lab results
with all kinds of helpful information: including platelet clumping,
microorganisms, RBC and WBC morphology, and loads more. What gives?
Hold on to your lab coats. I’ll get there in a minute.
Slide review and differential training in medical school
and residency
[This section intentionally left blank]
Image 2. There is nothing wrong with your television set. Do not attempt to adjust the picture. You are about to experience the awe and mystery which reaches from the inner mind to… the bench tech working in hematology. The one who went to school for this? Medical school and residency are starkly devoid of any in-depth, comprehensive learning for differentials.
A Differential, Differential
So let me address the issue I brought up: why do
hematologists come down to the lab to look at the slides themselves, when
perfectly capable BOC certified, degree-holding medical laboratory scientists
and pathologists sign out validated differentials? It might not happen this way
at all hospitals, but I think the answer is a simple two-part problem.
First, as with the many things I’ve learned in medical
school, one of the lab-centric pieces of information that is well understood is
that, well, no one really knows what the lab does and how it operates.
Virtually nobody knows the depth and breadth of the testing that pathologists
manage, let alone the scientific precision and accuracy that instrument
validation requires. Learning that MLS techs are certified, can hold graduate
degrees, and even do their own research is often surprising to most of our
clinical colleagues. And—I will tell you for a fact—that pathology and
laboratory testing methodology is not covered in medical school the way you
might think. Pathology is more of a class of distinguishing the identifying
details of a disease, not understanding the interdisciplinary diagnostic
teamwork that goes into those CBC index results on a computer screen on the
clinical floors.
Second, hematologists are specialists just like any other
practicing clinician. They know their stuff! They manage patient diagnosis,
treatment, and follow-up with the most up to date literature, national cancer
guidelines, and anything else available to better their patients’ outcomes. Despite
the notes in the CBC results that there are numerous macrocytes with
hypersegmented neutrophils, or 3+ schistocytes reported in a manual
differential—seeing is believing. It helps to see the slide yourself and get a
feel for the disease “state” with your own eyes. Moreso, it could be a learning
opportunity. It’s well within a clinicians’ scope to come down and look at a
peripheral smear, I actually encourage it. But it should come with a few
caveats…I’ll get to those too…
I-CARE
One of the places I was proud to hang my lab coat was
actually my first job as an assistant lab technician in the blood bank at Rush
University Medical Center in Chicago. Before I got my MLS and way before grad
school or med school, I was a blood bank “expediter.” Super fancy title, but
all I did was make sure specimens were logged in and blood products were up to
par with labels on their way out. Clerical but critical! (Let me have this,
please…haha) Anyway, part of the culture at that hospital has stayed with me
all these years. I’ve talked before about culture and the
way it permeates an institution’s practice like at the Mayo Clinic, but for
my first foray into clinical work their acronym was clutch: I CARE.
I for innovation
C for collaboration
A for accountability
R for respect and
E for excellence
Why am I telling you this? No, there are no royalties. I
just think it’s an easy way to remind ourselves about the meaning of
interdisciplinary medicine and they way we should work together across
specialties, and from bench to bedside. When we incorporate those values into
our work for the purpose of improving patient care and outcomes, everyone wins.
In this case, effective utilization of resources tells us that peripheral slide
review means different things to different people. In the setting of
hematologic work-ups, flags and review at the bench can signal something to the
clinician which could spark a conversation with the pathologist. All parts
contributing to a whole of patient care. Vigilante medicine is bad news.
Collaboration is key.
One place I was lucky enough to be a part of this
interdisciplinary collaboration was Swedish Covenant Hospital. One of the
hematology physicians would routinely call me and ask to look as peripheral
smears down in the lab, often as a group with med students, residents, and
fellows. I’d throw the image of his patients’ slides on a large flat screen and
go over what certain traits meant with regard to morphology and identification
from the lab setting. Dr. Cilley would add what this all meant clinically and
discuss treatment algorithms and next steps. That was collaboration at it’s
finest: lab tech working with pathologists, clinicians working with the lab,
and patient’s benefiting from all of it.
Video 1. ASCP’s 2015 Membership Video. I was super thrilled to be part of this video back in 2015 after winning the Midwest regional ASCP member of the year. If you’re bored enough to make it about 40 seconds into the video, that was my actual desk where Dr. Cilley and his residents would come to discuss patient slides. I would talk to them about morphology and hematologic clues with digital hardware and software to make it clear in group settings, rather than taking turns at the scope. Good times. (Source: https://www.youtube.com/watch?v=86fBRXGrZFo)
Video 2. Dr. Jeffrey Cilley talks about treating cancer as a “team approach” and he’s right. Hematology/Oncology to patient. Lab to clinician. Bench to bedside. (Source: https://www.youtube.com/watch?v=q0waKLyT1Dg)
Teamwork makes the dream work
About those caveats for collaboration I mentioned earlier…
Let me put it briefly: it’s well within the scope of a clinician to come over
to the laboratory and get some information on their patient’s lab
results/testing. But why not consider the following:
If a physician calls to review a smear, offer
to go over it with them. Likewise, to our clinical friends: if you go to the
lab for a slide don’t be batman—ask the tech what they think!
Experienced techs are one of the hospital’s most valuable resources. Some
folks I’ve worked with have been looking at slides longer than I’ve been using
my eyes at all! They’ll save you and your residents the time when those
terrifying intracellular microorganisms are really just overlying platelets. I
mean, they’ve got a cute halo.
If you need help, just ask. This applies to
everyone.
Talking with the tech about the slide is great start, but there’s more
resources in the lab than most people know what to do with! Clinical
physicians: check the shelves around the hematology microscope. Stuck on
something? Find a CAP atlas or a proficiency survey booklet guide. Easy to
read. Techs and pathologists: have someone who constantly comes down for slide
review despite your immaculate and detailed SOPs on CBC results reporting? Have
a quick chat about the work that goes into resulting those diffs—you might even
improve your heme TAT, who knows?
If it’s well within the right of a physician
to leave the unit and see a patient’s slide, logic says that maybe, just maybe,
it should be okay for a pathologist to leave the lab and see a patient at the
bedside!
Hospitals
are full of never-ending rounding white coats, all asking patients questions,
and all contributing specialty notes to their charts. But its not only
to prevent patients from getting a decent nap. We’re all parts of a large
interdisciplinary patient team. A recent Medscape survey found that somewhere
around 3% of pathologists see patients, routinely! Got an interesting case in
the lab, someone who’s part of lots of tumor boards, someone with an
interesting case to write up, or even someone who nobody knows exactly what’s
going on with? Try walking over to 4 south and have a conversation with Mr.
Jones; it might help. At least he’ll know how many people are working on his
care team!
The bottom line: we’re in this together, and like the flag
on the ASCP ship says, we’re Stronger Together. Innovation,
collaboration, accountability, respect, and excellence are—and should be—simple
cornerstones of clinical medicine that translate across every discipline. When
we share information and expertise, everyone gets better at what they do.
Bonus Image. This was a hard picture to take. Usually, a quick hematologist just comes down to see if there are any real schistocytes. But, after reading a draft of this post, BatDoc’s cool with chatting about red cell indices and automated flow cytometry methods in auto-diff validation. That’s the hero we deserve, and the one healthcare needs! (Source: https://gunaxin.com/batman-doesnt-police-stop-visiting-children-hospital)
Thanks for reading!
See you next time!
–Constantine E. Kanakis MSc, MLS (ASCP)CM graduated from Loyola
University Chicago with a BS in Molecular Biology and Bioethics and then
Rush University with an MS in Medical Laboratory Science. He is
currently a medical student actively involved in public health and
laboratory medicine, conducting clinicals at Bronx-Care Hospital Center
in New York City.
When a complete
blood count (CBC) and differential is ordered by a physician, most labs today
have instrumentation capable of performing an automated differential. Depending
on the instrument results and flags, we may need to perform a scan, review of
the slide, or a manual differential. However, the definition of a manual
differential today may be a bit different than the historical definition. A
typical manual differential, when I first started working as a technologist,
consisted of counting and differentiating 100 white blood cells under a
microscope, and performing a red blood cell morphology along with a platelet
estimate. Today, the 3 components of the manual differential have not changed,
but more and more labs are using an
automated digital counting device, such as CellaVision. Whether counting cells
under the microscope or scanning and verifying or reclassifying cells in CellaVision,
it is important to always address all 3 parts of the manual review.
When
an automated
CBC has flagged that abnormal RBC morphology may be present, a peripheral blood
smear should be reviewed. Reporting the red blood cell (RBC) morphology is an
important component of a differential. Evaluation and interpretation of RBC
morphology may provide the physician with important diagnostic information
regarding the underlying cause of a variety of disorders, including anemia and
systemic disease. Therefore, it is important to be able to accurately recognize and
identify RBC morphologic abnormalities.
Red blood cell
morphology can be subjective, and therefore inconsistent. Therefore, Laboratories
must have training and competency programs as well as procedures which dictate how they will report
RBC morphology. Some labs use a numbering system, 1+, 2+, 3+, and others
report, ‘rare’,
‘few’, moderate’ or ‘many’. Some morphological, such as rouleaux, can just be
reported as present, with no quantified. Any method is acceptable, as long as
there is consistency in reporting.
When performing
RBC morphology, these semi-quantitative
report formats for should be based on clinical significance. Some RBC
morphologies and inclusions are clinically significant,even when
they are present in very low numbers. Sickle cells are one of these
abnormalities that are significant even if only seen in very small numbers.
Malaria or other parasites are clinically significant in any number. Fragmented
cells such as schistocytes and helmet cells should also be noted if seen in any
number. Other abnormalities which can be clinically significant in very low
numbers are polychromasia, spherocytes and teardrop cells.
There are many
other abnormal RBC morphologies which are only clinically significant if seen
in larger numbers. Laboratories may choose to only report the presence of
ovalocytes, target cells, burr cells, macrocytes, microcytes or hypochromia
when greater than a defined percentage of cells exhibit these morphologies.
Other laboratories choose to not report macrocytes, microcytes and hypochromia
at all, instead relying on the physician to use the RBC indicies for their
indication. The 2 most important things to remember, whatever your procedures
are, is to be consistent, and not to ignore the:RBC morphology.
In
addition to performing RBC morphology, a manual differential also requires
platelet examination. A smear should be examined for a platelet estimate and
abnormalities. This is particularly important when platelet clumps or an
abnormal platelet scattergram are flagged on the CBC. If an instrument uses optical platelet
counts, large platelets can be missed. A fluorescent platelet count (PLT-F) ,
performed on Sysmex analyzers, will stain only platelets and give an accurate platelet
count. The fluorescent count eliminates interferences seen with other methods.
However, even when reporting a PLT-F, it may still required to review the smear
for a platelet estimate, particularly with a very low count, or with clumped
platelet flags. Clumped platelets are not an uncommon phenomenon, and an
accurate platelet count can not be reported if significant clumping is present.
The presence of giant platelets or hypogranular platelets, seen on the
slide, can also aid the physician in
diagnosis or patient management.
CellaVision users have the added benefit of automation which simplifies the process of performing manual differentials. The system automatically locates and takes digital images of cells, including white blood cells, red blood cells and platelets.This simplifies the process of performing a manual differential. White blood cells are pre-classified, RBC images are provided, and platelet images allow platelet estimates to be performed easily. The new advanced RBC application software can pre-classify RBCs. This makes it even easier than before to perform reliable, standardized RBC morphology. (Watch for my next Hematology blog about the new RBC software!)
Particular
disorders or abnormalities often involve characteristic changes to RBC
morphology “Assessment of RBC morphology can be the best tool for
laboratory hematology professionals to recommend clinical and laboratory follow‐up in a patient with anemia and to select the
right tests for definitive diagnosis.”1 Too
often, I have seen technologists perform a manual differential and either
superficially skim over the RBC and platelet components, or totally forget
them. Don’t forget
your RBC morphology and platelet estimate and morphology! With today’s
automated differential and autovalidation, 75-85% of CBCs are autovalidated.
This allows us to spend quality time on those manual reviews that need to be
done. Be sure to spend your time thoroughly reviewing the slides. A scan, slide
review or manual differential, whether done under the scope of with CellaVision,tells
the physician that we have looked at the slide or cells, which must include all
3 parts of manual review… WBCs , RBCs and platelets. Don’t sign it out until
it’s complete!
References
J. Ford, Red Blood Cell Morphology.
International Journal of Laboratory Hematology. 2013
-Becky Socha, MS, MLS(ASCP)CM BB CM graduated from Merrimack College in N. Andover, Massachusetts with a BS in Medical Technology and completed her MS in Clinical Laboratory Sciences at the University of Massachusetts, Lowell. She has worked as a Medical Technologist for over 30 years. She’s worked in all areas of the clinical laboratory, but has a special interest in Hematology and Blood Banking. When she’s not busy being a mad scientist, she can be found outside riding her bicycle.
Caster Semenya celebrates as she wins gold in the women’s 800 meters in the Commonwealth Games on April 13, 2018, on Australia’s Gold Coast (1). Jason O’Brien/Getty Images
I will continue this month along the thread of last month’s
post, which addressed the controversy surrounding South African female
mid-distance runner Caster Semenya. Caster has won many international
mid-distance races (400-800m), but she has been suspected of naturally
producing higher levels of testosterone.
Since last month, I’ve learned the reason for the higher
testosterone is uncertain: it could be due to natural production
(hyperandrogenism) or rumors of her being intersex1. Regardless,
what I will discuss here is how the proposed actions of the International
Olympic Committee would be expected to affect Semenya’s performance.
Specifically, how would lowering testosterone levels affect her athletic
performance?
Last month, we saw that muscle mass might be expected to
decrease, but this may not affect athletic performance significantly.
Another important effect of testosterone is on red blood
cell levels including hemoglobin, which by carrying oxygen to muscle is a
central part of calculating VO2max. VO2max is maximal oxygen
consumption. This is strongly linked to performance in cardiovascular athletic
events.
Mid-distance running requires a large cardiovascular
capacity. Maybe not the same level of Tour-de-France long distance bikers in
the Alps, but still substantial. As a runner that feels pretty proud at having
run a sub-3 minute 800m, I can say Caster’s feat of running it in less than 2
minutes is incomprehensible. From the burning feeling in my lungs and thudding,
maximum heart rate at the end of the half-mile, I can attest that this event
requires substantial cardiovascular efficiency.
Maximal oxygen consumption (VO2max) by exercising
skeletal muscle is principally limited most by cardiac output and
oxygen-carrying hemoglobin levels. This has been shown quite convincingly in a
series of experiments in the 1950’s-70’s2,3 that probably wouldn’t
be approved by the IRBs of today charged to protect research subject rights.
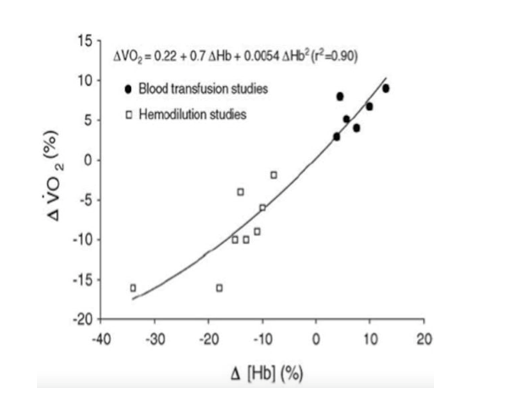
First, transfusing blood increased hemoglobin concentration
and similarly the VO2max and exercise endurance of participants. (This practice was exploited most notably later
on in the Tour de France). In other
studies3, blood was removed from participants before assessing their
exercise tolerance (10% loss of hemoglobin à
13% reduction in VO2max). Another study removed 400mL, 800mL and
1,200mL over several days, which decreased hemoglobin by 10%, 15%, and 18%
respectively. There was a concomitant decrease in endurance time (-13%, -21%,
-30%) and VO2max as well (-6%, -10%, -16%)3. A summary of blood transfusion and
hemodilution studies is shown in Figure 1 from Otto JM et al4.
Figure 1. Reproduced from Otto JM et al (4)
In transgender women (gender incongruent with sex assigned
male at birth), hormone therapy to increase estrogen levels (oral estradiol)
and block testosterone (anti-androgen: spironolactone) reduces hemoglobin by 9%
on average (from 15.2 g/dL to 13.9 g/dL)5. I would expect a smaller
decrease for Semenya as she will likely not get a full dose hormone regimen
used for transgender transition and because her testosterone levels wouldn’t be
as high as biologic males’. However, she
would still be expected to have lower hemoglobin- similar to donating a half or
whole unit of blood. If hemoglobin decreased even just 5%, that could affect
her performance substantially when the difference between competitors boils
down to seconds in mid-distance races.
Arguably, forced blood donation could produce the same
effects as testosterone-lowering therapy. But it would be far too dramatic to
suggest something like bloodletting by the International Olympic Committee.
In the end, I don’t feel qualified to say what should be
done in this case. All I can say is that I don’t think lowering Caster
Semanya’s testosterone levels will have the intended effect of decreasing
muscle mass. On the other hand, it would decrease hemoglobin levels tempering
her performance. But who should determine the point where her hormone levels
should be? There is such a strong biologic connection between hormone levels
and physiology that manipulating them for athletic fairness could be akin to
playing puppeteer.
References
North, Anna. ““I am a woman and I am fast”: what Caster Semenya’s story says about gender and race in sports” Vox. May 3, 2019
BALKE B, GRILLO GP, KONECCI EB, LUFT UC. Work capacity after blood donation. J Appl Physiol. 1954 Nov; 7(3):231-8.
Ekblom B, Goldbarg AN, Gullbring B. Response to exercise after blood loss and reinfusion. J Appl Physiol. 1972 Aug; 33(2):175-80.
Otto JM, Montgomery HE, Richards T. Haemoglobin concentration and mass as determinants of exercise performance and of surgical outcome. Extrem Physiol Med. 2013; 2: 33.
SoRelle JA, Jiao R, Gao E et al. Impact of Hormone Therapy on Laboratory Values in Transgender Patients. Clin Chem. 2019; 65(1): 170-179.
-Jeff SoRelle, MD is a Molecular Genetic Pathology fellow at the
University of Texas Southwestern Medical Center in Dallas, TX. His
clinical research interests include understanding how the lab intersects
with transgender healthcare and advancing quality in molecular
diagnostics.