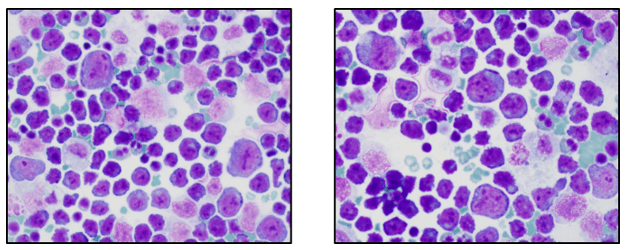
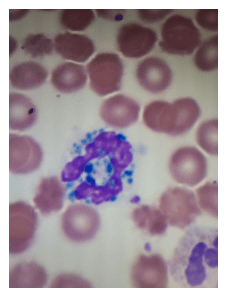
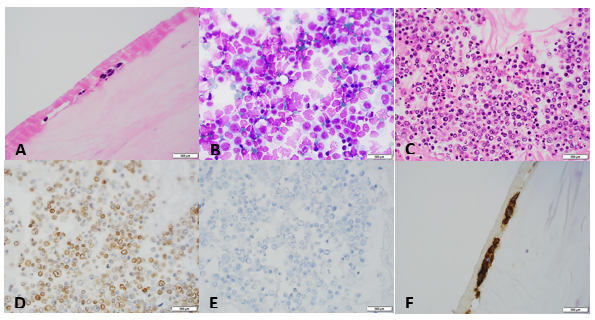
If you read my last blog, you heard the about the story “if You Give a Mouse a Cookie” by Laura Numeroff.4 The curious little mouse has a mind that never rests. As his mind wanders and hops from one thing to another, he keeps discovering more things to check out along the way. Medical laboratory lcientists are a lot like this. We’re a curious bunch, and, in investigations, one thing often leads to the next. Well folks, the mouse has struck again! We were given another cookie in the form of these beautiful cells.
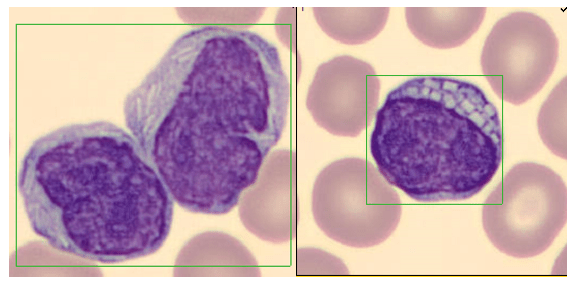
These cells were found by my coworker Liz Marr, MLS(ASCP), and the adventure began! First, we wanted to know what those were, and then we needed to find out more about them, and then, mostly, I wanted to know why in almost 40 years of working in and teaching hematology that I have never before seen this!
The story begins with our case history. We received a CBC from a 71 year old female with a 4 year history of untreated chronic lymphocytic leukemia/ small lymphocytic lymphoma(CLL/SLL). The patient’s recent history included a myocardial infarction(MI) 5 months prior. The patient was found to have leukocytosis (WBC 25.38 x 103/μL) and absolute lymphocytosis (18.25 x 103/μL) with normal hemoglobin and hematocrit (Hgb 13.4 g/dL, Hct 40.8%) and normal platelet count (272 x 103/μL). The differential had 71.9% lymphocytes with many abnormal forms noted. The slide was sent for a pathology review. The pathologist reported “Atypical lymphocytosis consistent with patient’s known chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) Filament-like inclusions are present in the cytoplasm which has been previously reported in patients with CLL.”

A curious tech can’t stop at just that description. If you tell me they are filament-like inclusions, I will have all kinds of questions. What are these filaments made of? Are they crystals, or something else? How common are these? Are these diagnostic of CLL? Are these only seen in CLL? What is their significance? And, of course, the most puzzling question, why have I never seen these before??
CLL is a form of non-Hodgkin lymphoma and is the most common leukemia in the Western world. It is generally a leukemia of older age with a median age at diagnosis around 67-72. The disease is widely variable, with some patients asymptomatic and requiring no treatment for many years, while others have a more rapidly progressive course of disease requiring treatment. About 60% of patients are diagnosed before they exhibit any symptoms. CLL and SLL are considered to be different manifestations of the same disease. In CLL, the abnormal B lymphocytes are found mostly in the peripheral blood and bone marrow, but in SLL, there is lymph node involvement, with abnormal cells mostly found in the lymph nodes. CLL is diagnosed based on absolute B lymphocyte counts ≥5 x 109/L. Flow cytometry typically reveals a distinctive cell immunophenotype with expression of CD19, CD5, CD23, and Κ/λ; and weak expression of CD20, CD79b, and surface immunoglobulin.1
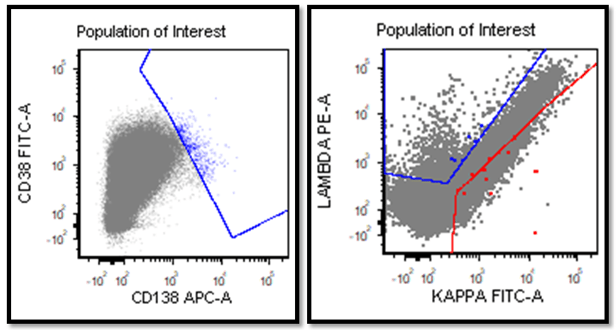
The most recent flow cytometry report on our patient was from one year ago. An 8 color analysis with CD45/SSC gating was performed by LabCorp. The flow revealed an abnormal cell population representing 56% of total cells. Two monoclonal B cell populations were detected with identical phenotypes except for light chain expression. These cells expressed CD45, CD19, CD20, CD22, CD5, and CD23., CD38-. This phenotype was consistent with her previous diagnosis of CLL/SLL.
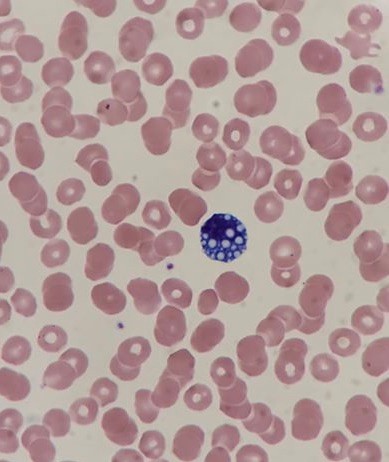
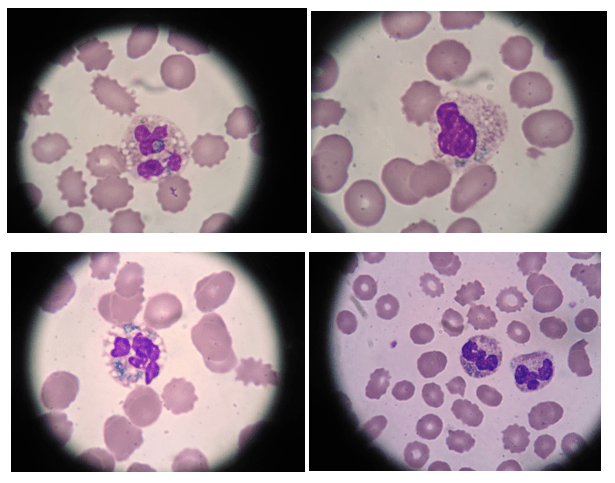
A literature search revealed only a few articles about intracytoplasmic inclusions in CLL. Cytoplasmic inclusions in lymphomas are uncommon, but have been noted as vacuoles, crystals, and pseudocrystals. These crystalline inclusions represent immunoglobulin(Ig) heavy and light chain that precipitate in the cytoplasm. Using electron microscopy it has been found that theses Ig deposits localize in the rough endoplasmic reticulum (RER).5 When surface Ig can be demonstrated on the B lymphocytes, it has been found to be same as Ig in the inclusions.6
In two published studies that describe these crystal like inclusions, photographs are very similar to the ones we found on our patient.3,5 It is interesting to note that, in these two studies, neither of the subjects was a known CLL patient. The inclusions were noted in the patients’ cells and the peripheral blood was subsequently sent for flow. Phenotypes reported confirmed monoclonal B-cells representing a large percentage of cells. Huang reported monoclonal B-cells which expressed CD45, CD19, CD20, CD22, CD79b, CD5, CD23, CD148 and CD200(hi), with partial expression lambda, and negative for FMC7, CD10, CD11c, CD49d, CD103, CD38, CD25, CD160, IgM, CD81, kappa and Ki67.3 In the Ramlal case study, phenotype was CD5, CD19, CD20, CD23, positive, CD10, FMC7 negative.5 On the basis of flow, along with the CBC results, the patients were diagnosed with CLL.
Of course, while researching this, the little mouse in me kept asking questions and finding more questions to ask. One question that I still had questions about was if these inclusions have any prognostic value. In three recent studies3,5,6 it was indicated that these inclusions can be used to help with diagnosis, but are not prognostic for course of disease. Rodriguez followed a patient with asymptomatic Rai stage 0 CLL. This patient consistently had inclusions noted in lymphocytes for 9 years before any progression of disease was noted.6 In the medical field even if one study reports no prognostic significance, this opinion could change in the future with more studies. Could these crystalline inclusions be used to forecast time to first treatment (TFT) or overall survival?(OS). So far, because of the rarity of these cytoplasmic inclusions, there is no evidence of prognostic value. As well, the mechanism related to their formation and their role in CLL is yet to be determined.
Our case study patient and the various reports found in literature had common flow cytometry immunophenotypes. Patients were all either previously diagnosed with CLL or lymphocytic lymphoma, or were diagnosed at the time of the findings of these inclusions. While these crystalline inclusions alone are not considered diagnostic for CLL, their recognition can be used to assist in a prompt diagnosis of a lymphoproliferative disease. And they are so pretty! What medical laboratory scientist doesn’t love pretty cells? Be like that mouse. Be curious, keep your eyes open, and be on the lookout for these interesting cells in CLL patients, but, more importantly, in patients with lymphocytosis without a known diagnosis of a lymphoproliferative disorder.
References
- AJMC, January 7, 2019
- Chronic Lymphocytic Leukemia: An Overview of Diagnosis, Prognosis, and Treatment
- Huang, Y., Zhang, L. Intracellular rod-like crystals in chronic lymphocyte leukemia. Int J Hematol 112, 267 (2020). https://doi.org/10.1007/s12185-020-02933-7
- Numeroff, Laura If You Give a Mouse a Cookie. 1986
- Ramlal, B, DiGiuseppe, JA. Intracytoplasmic crystalline inclusions in chronic lymphocytic leukemia. Clin Case Rep. 2019; 7: 1460– 1461. https://doi.org/10.1002/ccr3.2250
- Cecilia M. Rodríguez, Carmen Stanganelli, Claudio Bussi, Daniela Arroyo, Darío Sastre, Viviana Heller, Pablo Iribarren & Irma Slavutsky (2018)Intracytoplasmic filamentous inclusions and IGHV rearrangements in a patient with chronic lymphocytic leukemia, Leukemia & Lymphoma, 59:5,1239-1243, DOI: 10.1080/10428194.2017.1370549

-Becky Socha, MS, MLS(ASCP)CM BB CM graduated from Merrimack College in N. Andover, Massachusetts with a BS in Medical Technology and completed her MS in Clinical Laboratory Sciences at the University of Massachusetts, Lowell. She has worked as a Medical Technologist for over 30 years. She’s worked in all areas of the clinical laboratory, but has a special interest in Hematology and Blood Banking. When she’s not busy being a mad scientist, she can be found outside riding her bicycle.