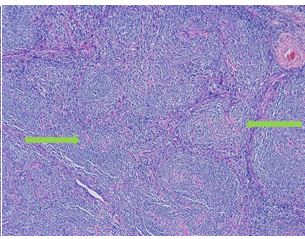
Lymphoid neoplasias are traditionally categorized as Hodgkin Lymphomas or Non-Hodgkin Lymphomas. Hodgkin Lymphomas are typically characterized by both expansive reactive lymphocytes and a paucity of the interspersed, malignant Reed-Sternberg (RS) Cell. These RS cells are large neoplastic B-Cell variants with ‘owl-eye’ nucleoli within multilobed nuclei. The background reactive lymphocytes are non-neoplastic cells drawn by secreted RS cytokines (IL-5, IL-6, IL-13, TNF, and GM-CSF), which often result in presenting B symptoms (fevers, chills, and night sweats). Histiocytes, granulocytes, and plasma cells are also commonly identified. Confirmatory immunohistochemistry staining for RS cells involves CD30 positivity, CD15 positivity, and negativity for CD20 and LCA (CD45) staining. The two major types of Hodgkin Lymphoma are recognized as Nodular Lymphocyte Predominant Hodgkin Lymphoma (NLPHL) and Classic Hodgkin Lymphoma (CHL). In NLPHL, B-Cell immunophenotype is generally preserved and is histologically recognized by a nodular predominance of small lymphocytes and RS cell variants, called lymphocyte predominant (LP) or popcorn cells (formerly called L&H cells for lymphocytic and/or histiocytic RS cell variants), which exhibit muted, lobed nuclei and smaller nucleoli. The Classic Hodgkin Lymphomas are categorized histologically as Nodular Sclerosis CHL (NSCHL), Lymphocyte Rich CHL (LRCHL), Mixed Cellularity CHL (MCCHL), and Lymphocyte Depleted CHL (LDCHL).
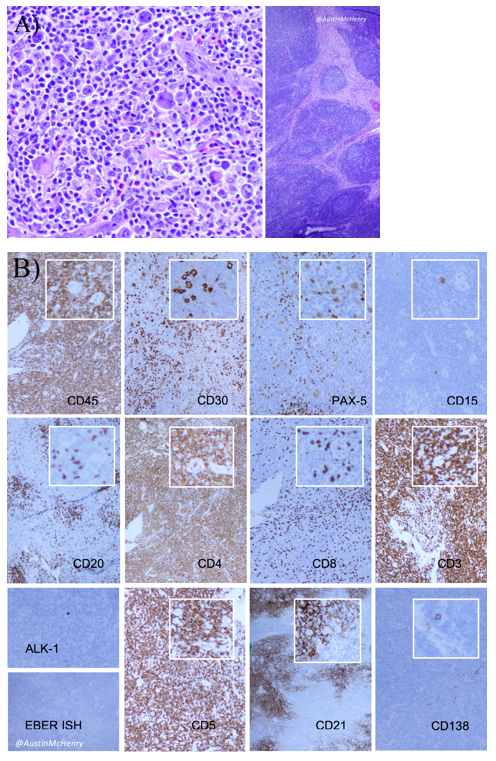
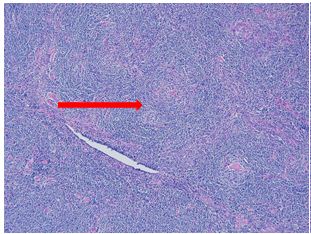
Nodular Sclerosis Classic Hodgkin Lymphoma (NSCHL) comprises over 70% of all CHL and is characterized by broad, fibroblast-poor collagen banding surrounding at least one nodule and by RS cells with lacunar morphology. The RS cells tend to have a larger amount of cytoplasm than those in other types of classic Hodgkin lymphoma. When fixed in formalin, this excess cytoplasm causes their membranes to retract so that the cells seem to be sitting in lacunae. Typically, EBV association is uncommon, CD30 is almost always expressed, CD15 is expressed in 75-85% of cases, and PAX5 is weakly positive. Most cases do not express T-Cell antigens (CD4, CD3, CD8), but when they do (~5% of cases), they can be difficult to distinguish from ALK-negative Anaplastic Large Cell Lymphomas (PAX5 positivity can help to rule out ALCL). True T-cell marker expression on RS cells has been seen (usually weak) and is associated with a poorer prognosis.
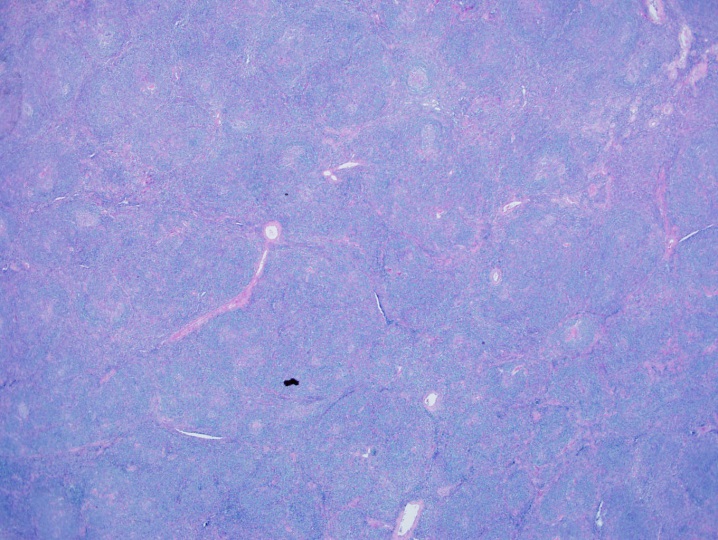
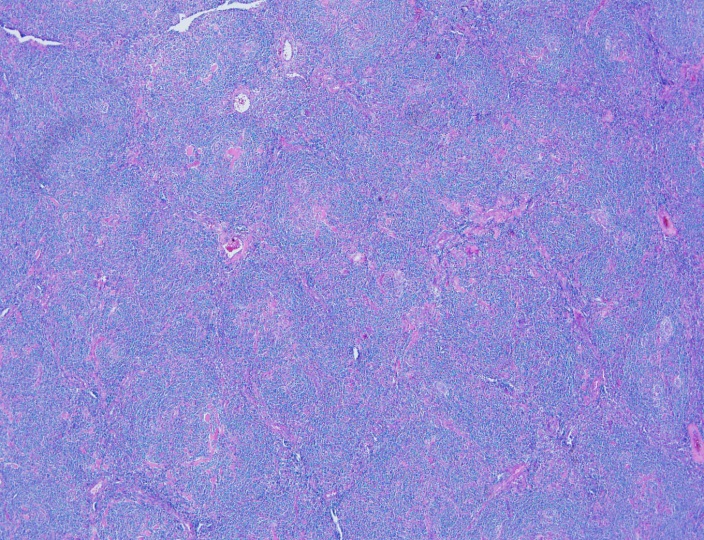
Two histologic variants of NSCHL have been proposed: a fibrocystic variant (FV) and a syncytial variant (SV). The FV contains abundant fibroblasts and histiocytes in the setting of difficultly identified RS cells. The SV describes the finding of RS cells in prominent, sheet-like aggregates forming cohesive lacunar nests in the center of nodules (Figure 1). The SV represents 5-16% of all cases of NSCHL (Ben-Yehuda-Salz et al. 1990), though some suspect it may be as high as 25% (Sethi, T., et al. 2017).
Advancements in chemoradiation therapies have improved the long-term survival of patients of with CHL. Today, more than 85% of patients with early stage CHL will be cured of the disease (Ansell, S., et al. 2014). The 5-year overall survival for early stage CHL is 95% and for advanced disease is 82%. Overall, the prognosis of NSCHL is better than that of other types of CHL.
Initial studies examining the prognostic significance of grading of NSCHL based on cytological and tumor microenvironment features showed an association of higher grade (based on the British National Lymphoma Investigation (BNLI) criteria (Bennett et al. 1981; MacLennan et al. 1989)) with a poorer prognosis (MacLennan et al. 1989; Wijlhuizen et al. 1989; Ferry et al. 1993). However, since the advent of combination chemotherapy and the relatively good prognosis of NSCHL today, the significance of such grading or further classification has been called into question. Importantly, grading based on proposed histological features has declined in the last two decades because such advances in therapy can actually obscure the differences seen in less-effectively treated patients. As such, grading is not currently deemed necessary for routine clinical purposes.
Despite the broadly recognized clinical favorability of NSCHL, slowly accumulating evidence of poor clinical outcomes associated with the syncytial NSCHL histological variant is only now beginning to declare itself with strong evidence. While BNLI grading may no longer be a useful clinical tool, variant typing may instead prove effective as both a prognostic and alternative treatment indicator.
Small case studies have reported aggressive disease in SV NSCHL since the first histologic case was described. Clinical behavior has included presenting with a considerable mediastinal mass, significant B symptoms, and advanced stage at diagnosis. Despite these case reports, it was not until recently that systematic analysis of progression free survival and complete therapy response among morphology and immunophenotype SV confirmed cases in patients with similar treatment regimens (combination doxorubicin, bleomycin, vinblastine, and dacarbazine, or ABVD) was conducted (Sethi, T., et al. 2017).
Sethi, T., et al evaluated 167 patients with NSCHL: 43 patients with SV were compared with 124 patients with typical NSCHL on patient characteristics, disease variables, treatment administered, and outcome at Vanderbilt University Medical Center between 1995 and 2014. The rate of complete treatment response was lower in the SV variant as compared with typical NSCHL with standard induction therapy, 74% versus 87% (p= 0.05). Patients with SV had a shorter progression free survival and experienced disease relapse. The median progression free survival for the entire cohort was 174.7 months. The median progression free survival in the SV group was 17.02 months which was significantly shorter compared with that of the typical NS group, which was not reached (p < 0.0001).
The BNLI criteria for the grading of NSCHL (Grades I-II) are based on the amount of sclerosis, the degree of nodular cellularity, and the number of atypical neoplastic cells. Most SV cases are grade II because of the number of atypical cells present. However, most grade II cases are not syncytial variants. Therefore, BNLI grade alone is not an accurate depiction of the clinicopathologic features of this disease. In the same way, it may also not be the best determinant of disease prognosis, therapeutic indication, or investigative classification.
Patients with Syncytial Variant Nodular Sclerosis Classic Hodgkin Lymphoma experience a lower than expected rate of complete therapeutic response with shorter progression-free than non-SV NSCHL treated with standard therapy. Syncytial Variant NSCHL should therefore be recognized as a high-risk subgroup within the otherwise traditionally docile NSCHL classification. It is time the SV finally be considered a true histopathologic variant in future trials involving novel agents to assess treatment response. While a majority of patients with SV NSCHL can likely be successfully salvaged with high-dose therapy and autologous stem cell transplantation, studies of novel agents such as conjugated antibodies or immunotherapeutic agents should be considered in these patients to improve complete response rates and to avoid the need for toxic salvage therapies.
References
- Sethi T., et al. Differences in outcome of patients with syncytial variant Hodgkin lymphoma compared with typical nodular sclerosis Hodgkin lymphoma. (2017) Ther. Adv. Hematol. 8(1):13-20.
- Granot, N., et al. Syncytial variant of nodular sclerosing Hodgkin lymphoma in children: A prognostic factor? (2018) J. Ped. Hem. & Onc. 35;1:33-36.
- Ansell, S. (2014) Hodgkin lymphoma: 2014 update on diagnosis, risk stratification, and management. Am J Hematol 89: 771–779.
- Bennett, M., Maclennan, K., Easterling, M., Vaughan Hudson, B., Jelliffe, A. and Vaughan Hudson, G. (1983) The prognostic significance of cellular subtypes in nodular sclerosing Hodgkin’s disease: an analysis of 271 non-laparotomised cases (BNLI report no. 22). Clin Radiol 34: 497–501.
- Ben-Yehuda-Salz, D., Ben-Yehuda, A., Polliack, A., Ron, N. and Okon, E. (1990) Syncytial variant of nodular sclerosing Hodgkin’s disease. A new clinicopathologic entity. Cancer 65: 1167–1172.
- Ferry, J., Linggood, R., Convery, K., Efird, J., Eliseo, R. and Harris, N. (1993) Hodgkin disease, nodular sclerosis type. Implications of histologic subclassification. Cancer 71: 457–463.
- Maclennan, K., Bennett, M., Tu, A., Hudson, B., Easterling, M., Hudson, G. et al. (1989) Relationship of histopathologic features to survival and relapse in nodular sclerosing Hodgkin’s disease. A study of 1659 patients. Cancer 64: 1686–1693.
- Wijlhuizen, T., Vrints, L., Jairam, R., Breed, W., Wijnen, J., Bosch, L. et al. (1989) Grades of nodular sclerosis (NSI-NSII) in Hodgkin’s disease. Are they of independent prognostic value? Cancer 63: 1150–1153.
- Strickler, J., Michie, S., Warnke, R. and Dorfman, R. (1986) The “syncytial variant” of nodular sclerosing Hodgkin’s disease. Am J Surg Pathol 10: 470–477.
- Van Spronsen, D., Vrints, L., Hofstra, G., Crommelin, M., Coebergh, J. and Breed, W. (1997) Disappearance of prognostic significance of histopathological grading of nodular sclerosing Hodgkin’s disease for unselected patients, 1972–92. Br J Haematol 96: 322–327.
- Hess, J., Bodis, S., Pinkus, G., Silver, B. and Mauch, P. (1994) Histopathologic grading of nodular sclerosis Hodgkin’s disease. Lack of prognostic significance in 254 surgically staged patients. Cancer 74: 708–714.
- Darabi, K., Tester, W., Daskal, I. and Cohn, J. (2015) Syncytial variant of nodular sclerosing Hodgkin’s disease. Blood 104: 4533–4533.

-Austin McHenry is an M3 at Loyola University Stritch School of Medicine in Maywood, IL. Austin is past-president of the pathology intrest group SCOPE (Students Curious about Outrageous Pathology Experiences) and is a recent recipient of ASCP’s Academic Excellence and Achievement in Pathology Award. Follow Austin on Twitter @AustinMcHenry

-Kamran M. Mirza, MD PhD is an Assistant Professor of Pathology and Medical Director of Molecular Pathology at Loyola University Medical Center. He was a top 5 honoree in ASCP’s Forty Under 40 2017. Follow Dr. Mirza on twitter @kmirza.