Pre-analytic factors contribute to over 70% of laboratory errors. The most common examples include missing test request, wrong/ missing ID, contamination from collection site, hemolyzed/ clotted/ insufficient sample, wrong container or wrong transport conditions.
In our NGS lab, pre-analytic factors rarely arise. Sometimes an incorrect slide was sent, so then the tumor and germline DNA don’t match. We do perform many rounds of PCR (total 30-35 cycles), so contamination of amplified PCR products could be a concern. Usually, our volume is low enough that contamination isn’t a significant issue and not one we have noticed.
Multiple mutations- A COVID-19 Hybrid?
In a batch of COVID-19 whole genome sequencing samples from early July, I noticed some interesting mutations. Mutations from the Alpha and Delta variants were showing up in the same specimen. At first I thought this could mean a hybrid virus. Another possibility was co-infection in the same patient. And lastly, the specimen could just be contaminated. The best way to resolve this issue is to check the “phase” of the viral variant sequencing reads. The phase refers to the single pieces of DNA that are read by the sequencer. They are normally short (75-150bp in length), but if you can see whether the variants occur on the same read or only on different reads it rules out the possibility of a hybrid virus.

Determining the difference between a hybrid or 2 viral genomes
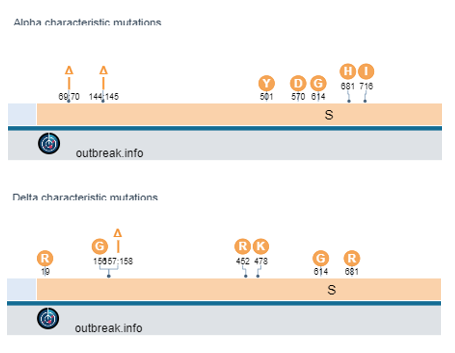
The difficulty is finding a location within both the Alpha and Delta variants that have mutations close to each other. One region exists near a deletion at amino acid 144 (Alpha) and amino acids 157-158 (Delta).

Tracing the source of the issue
As there were no overlaping reads with the 3 b.p. and 6 b.p. deletion, we concluded that this finding arose from 2 distinct viral genomes. As to whether the person was infected with both Alpha and Delta, we looked to the rest of our 96 well plate. A characteristic muation in Alpha spike protein is N501Y (it confers increased binding to ACE2R and increased infectivity). This mutation was found in several specimens of the Delta lineage, but in this case there was a much lower frequency of this variant compared to total reads. Also, several of the cases had high CT values to start with. Many of the contaminated samples also were near a variant that mapped strongly to Alpha and had a lower Ct value (CT=25). Mapping the location of the specmens to the plate showed close proximity to the authentic Alpha variant.
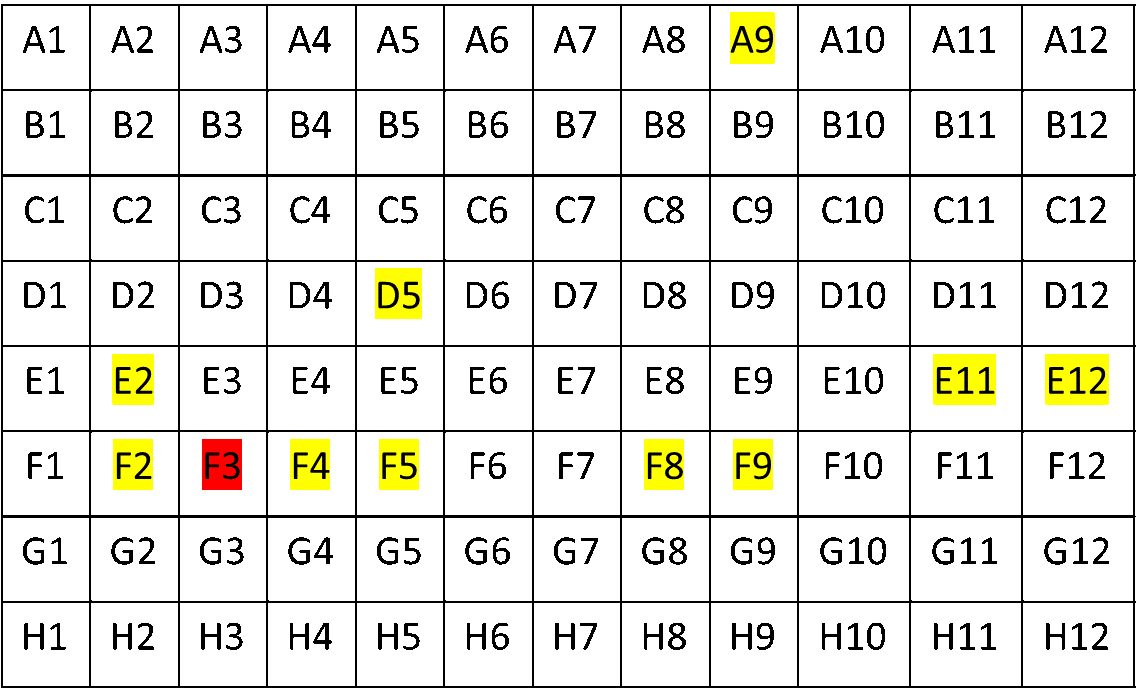
Luckily, before all WGS testing, we perform a targeted PCR, which I’ve mentioned in previous blog posts. This showed that only one sample (well F3) was B.1.1.7, and the rest were Delta variants. Thus we concluded we had experienced a case of contamination.
Since this time, we’ve had issues with the negative control having borderline positive levels of sequencing data that maps to Delta (now 100% of all cases and with low CT values). This is apparently a problem at multiple labs, but one that we are trying to address by looking at several root causes.
Pre-analytic concerns: Contamination sources
COVID-19 viral genome sequencing has found issues of contamination in several circumstances. We attribute this to a few reasons:
- High viral load of some specimens (especially Delta)
- Thin plastic covers that pop off easily, especially when taken out of the freezer
- And a large volume of specimens being processed.
For an example of the plastic cover issue, you can see this picture below where the cold temperature of -80C causes the plastic film adhesive to come off quickly as it is removed from cold storage. It makes popping sounds and could aerosolize viral particles to other wells in the plate.

We now use aluminum PCR plate covers that do not come off with freeze/ thaw transitions. Furthermore, we use a multi-channel pipette that pieces the cover to withdraw individual samples without exposing them to other wells.
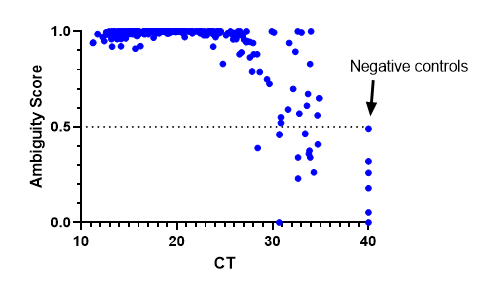
We have also implemented bioinformatic QC metric cut-offs to determine where a cut-off for negative specimens should be. We decided on an ambiguity score <0.5. The results were consistent with previous findings where lineages could be assigned at a CT >30, but sometimes they failed as CT values increased. Negative controls were assigned a CT of 40 and all fell below the 0.5 cut-off. This has been a useful metric to be sure we are providing high quality results.
Concluding remarks
Pre-analytic factors impact every part of testing and as COVID-19 sequencing has shown, even the NGS lab tests are not immune to these challenges.
The targeted PCR test helped flag/ resolve several of the issues as they arose.
COVID-19 sequencing is still for research/ epidemiologic purposes and demonstrates the importance of rigorous clinical validations to mitigate issues such as carry-over.
References Lippi G, Chance JJ, Church S, Dazzi P, Fontana R, Giavarina D, et al. Preanalytical quality improvement: from dream to reality. Clin Chem Lab Med. 2011;49:1113–26.

-Jeff SoRelle, MD is Assistant Professor of Pathology at the University of Texas Southwestern Medical Center in Dallas, TX working in the Next Generation Sequencing lab. His research interests include the genetics of allergy, COVID-19 variant sequencing, and lab medicine of transgender healthcare. Follow him on Twitter @Jeff_SoRelle.