Hello everyone, it’s been quite some time since my last post. I hope everyone has remained safe and healthy during these times!
My last post dived into short tandem repeat (STR) analysis for bone marrow engraftment monitoring. Today is a presentation of a patient who was transplanted for treatment of acute myeloid leukemia (AML). With all patients (with minor exceptions), donor and pre-transplant recipient samples are taken before transplant. Their informative alleles are then identified and used to determine the percent of donor and any recipient cells in subsequent post-transplant samples.
This patient was unique in that we were not able to obtain the donor sample (they were transplanted outside of our system), and therefore we used a buccal swab for their pre-transplant recipient informatives.
Buccal swabs are chosen because they are a non-invasive way to obtain squamous epithelial cells. These cells are important because they are of the recipient origin and will not change. With this technique, it is essential that the patient has no mucosal inflammation or is not too rough when swabbing their cheek. Otherwise, the buccal sample may become contaminated with blood which would contain donor cells.
We then inferred the donor informatives from the data of a mixed sample and the buccal swab.
Calculation of recipient and donor percentage in a post-transplant sample is determined on specific formulas that utilize these informative alleles. But what happens when a patient relapses and new mutations or deletions are introduced into their genome, causing a change in these informative alleles?
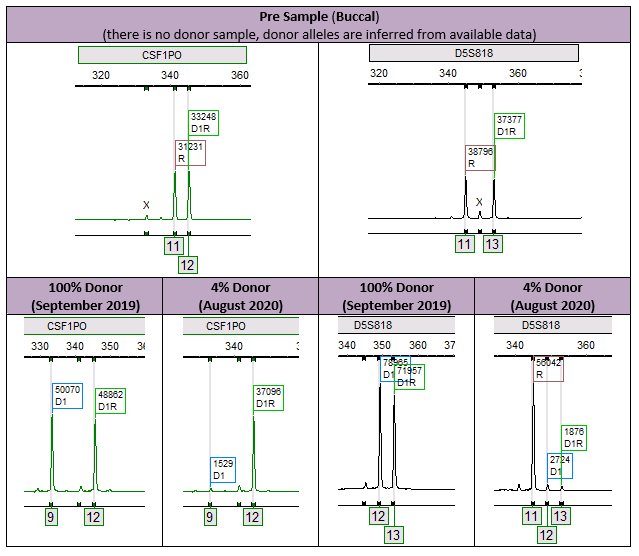
In this case, the patient had a loss of allele at two loci (CSF1PO – allele 11 and D5S818 – allele 13) after having previously obtained full engraftment (Figure 1).
Figure 1. The pre-sample was acquired through a buccal swab. There was no donor sample that was acquirable, and therefore the donor informative alleles were inferred through available data. In September of 2019, the patient was at 100% donor. Almost a year later, the patient is now at 4% donor and missing previously identified recipient alleles, indicating a loss of allele/mutation. Brown box with “R” stands for recipient. Blue box with “D1” stands for donor. Green box with D1R stands for shared.
The importance here is that the true percent donor is 4% (Figure 2). If we take a look at the affected informative alleles, we see an erroneous result of 100% donor and NI (which means the locus is non-informative, eliminating it from the calculations). This expands on the importance of an analyst to be attentive to the results presented. While this case was clearly evident and was caught by our error measurements, it is theoretically possible to cause an issue, especially in cases where the recipient percentages may be smaller. Furthermore, this phenomenon stresses the importance of including multiple informative alleles in our analysis, which increases our measurement of confidence.1
Figure 2. CSF1PO and D5S818 are incorrectly representing the patient’s status. CSF1PO is representing the patient at 100% donor and D5S818 is automatically identified as a non-informative by our software. After automatic and manual loci ignores, the total percent donor was 4%
We know that a loss of allele (loss of heterozygosity) is the likely explanation because both loci are in locations specific to the disease. Looking at Figure 3 below, the two alleles were affected because they were both present on the long arm of chromosome 5. Further, this chromosome is known to be involved in AML, and is also, of course, associated with other disorders like MDS.2 Additionally, the patient had cytology testing that identified this as an affected chromosome.
Figure 3. CSF1PO and D5S818 are both located on the long arm of chromosome 5. CSF1PO’s location is 5q33.1 and D5S818’s location is 5q23.2.
This is an interesting phenomenon and one that shows in measurable terms how a patient’s status can affect their molecular results. It’s further an expression of the molecular mechanisms of a disease, one of my first measurable experiences of how a disease affects the physical molecular constituents of another human.
To me, this encounter was an expression of how complicated, and yet connected, the entire genome has been designed. I am continuously amazed and look forward to expanding my understanding of molecular science.
References
Crow J, Youens K, Michalowski S, et al. Donor cell leukemia in umbilical cord blood transplant patients: a case study and literature review highlighting the importance of molecular engraftment analysis. J Mol Diagn. 2010;12(4):530-537. doi:10.2353/jmoldx.2010.090215
Crow J, Youens K, Michalowski S, et al. Donor cell leukemia in umbilical cord blood transplant patients: a case study and literature review highlighting the importance of molecular engraftment analysis. J Mol Diagn. 2010;12(4):530-537. doi:10.2353/jmoldx.2010.090215
-Ben Dahlstrom is a recent graduate of the NorthShore University HealthSystem MLS program. He currently works as a molecular technologist for Northwestern University in their transplant lab, performing HLA typing on bone marrow and solid organ transplants. His interests include microbiology, molecular, immunology, and blood bank.
Population-wide testing to identify symptomatic and asymptomatic infections could be a powerful tool to control Coronvirus Disease 2019 (COVID-19) spread, but current global testing capacity does not permit widespread testing of asymptomatic individuals. These tests are still limited to individuals who are symptomatic with limited availability to those with recent exposure to an infected person.
Because of the high prevalence of asymptomatic COVID-19 infections, proposals from the Rockefeller Foundation for disease mitigation include widespread and frequent testing of the US population. In the United States, diagnostic testing for SARS-CoV-2, the causative virus of COVID-19 is currently >2 million per week. Estimates for US testing needs for population wide surveillance range from 30 to 300 million per week. In order to scale testing by an order of magnitude, novel technologies and rethinking current testing paradigms are needed. The NIH has initiated a rapid funding program to develop SARS-CoV-2 testing, and these new technologies may play a part. However, we can broadly conceptualize key problems to address in population-wide testing in the US. The first is high-sensitivity testing which identifies active infection and can be performed with massive throughput. The second is the logistics of gathering hundreds of thousands of samples to each testing laboratory each day.
Next Generation Solutions to COVID testing
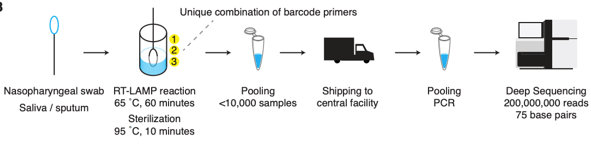
Emerging technologies using targeted next-generation sequencing have been suggested as a potential solution to population-wide testing. The key features include 1) extraction free amplification 2) an easily collected specimen such as saliva, 3) nucleotide barcodes to enable sample pooling, and 4) a limited number of targets (to allow deeper sequencing, i.e. higher sensitivity). Illumina is selling a whole genome test for SARS-CoV-2, but this limits sequencing to 3,000 tests/ run. Another recent approval for a private testing lab uses only one target, and may allow it to increase to 100,000 tests/ day. And a recent protocol for LAMP-Seq in pre-print outlines how this could work in a scheme below. An attractive aspect of this approach is decentralized specimen processing.
Whereas Bill Gates has supported a portfolio approach to vaccines placing multiple bets on different processes in parallel, a similar approach should be applied to multiplexed sequencing methods. Two sequencing runs can be performed on a single instrument in a single day, which can process several thousand samples. However, sequencing is not the only step in sequencing; library preparation and specimen handling take significant amounts of time too.
Laboratory Logistics
This technology would represent an exponential expansion in analytic testing capacity, but clinical labs will require a similar escalation in logistic capacity. The largest clinical laboratories in the world process less than 100,000 samples per day. Clinical laboratories have a long history of automation with the first robotic specimen track systems developed in the 1980s. Engineering and clinical lab expertise should thus partner to innovate on methods to handle high volumes. This level of investment for an issue that is likely to fade in 2 years, is not attractive to most private health systems, so public investment from multiple states in regional reference labs is needed.
It is still hard to conceive the necessary scale up in sample processing can be achieve within the time frame needed, so I would also propose a de-centralized sample processing approach. This would include self-collection of saliva (a safe, effective sample type with similar sensitivity as nasopharyngeal swabs), drop-off sites, and processing at places like Pharmacies (>90% of Americans live within 5 miles of a pharmacy and they could be authorized to administer tests- just as they administer vaccines). This would introduce pre-analytic problems, but if the goal is frequent and high rates of testing, then we will have to accept certain losses in sensitivity (which currently is arguably better than it needs to be). Interestingly, pre-analytic concerns with saliva have not led to sample instability or degradation of RNA causing false negatives, as described in my last post. However, other factors could affect saliva quality: smoking, age, and genetic factors of water: protein ratio affecting viscosity.
Testing solutions should be considered in the context of the planned testing network. The specimen type should be easy for the patient to provide, processed with existing laboratory equipment and resulted electronically. For example, current COVID-19 testing is based on sample collections requiring a healthcare worker encased in personal protective equipment (PPE) utilizing a swab device. Testing needs to progress to a simpler solution such as saliva which can be collected by the patient in the absence of a swab or PPE. Preliminary studies have demonstrated that saliva is sample type comparable to nasopharyngeal swab. The ideal saliva sample would be collected into an existing collection tube type (e.g. red-top tubes) which are already compatible with existing laboratory automation. In aggregate, a person could spit into a tube at-home, have the tube sent to a laboratory, and in the laboratory the tube would be directly placed onto an automated robotic track system.
Laboratory professionals need to provide a comprehensive plan for regional and national laboratory networks which can scale to provide overwhelming force to COVID-19 testing. No other profession or governmental organization understands testing as much as we do. Our understanding of managing samples from collection to result should be applied to the pandemic at hand. Until now most laboratorians in the US have focused on the immediate needs of providing testing for symptomatic patients and healthcare workers.
Vision for automated COVID-19 testing
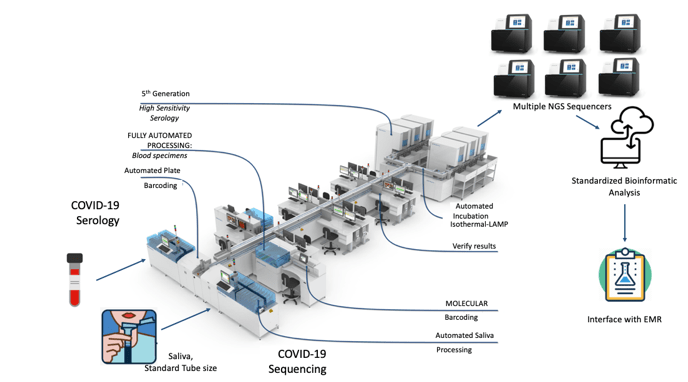
One could envision an automated line of testing that moves samples through processing to allow multiplexing and combinations of samples to allow large numbers of patients to be tested at once (see below). This is feasible in some specialized centers, but would require investments in automation, bioinformatics, and interfaces for a seamless process (figure below). If testing mostly asymptomatic patients, it may also be possible to do this on pooled samples. The number of samples to pool would depend on the likelihood to having a positive result (this would require sequencing all individuals in a pool).
This represents a synthesis of ideas in decentralized specimen collection, laboratory automation and massive testing throughput with Next-Generation Sequencing, but unfortunately this is not yet a reality.
References
Jonathan L. Schmid-Burgk et al. LAMP-Seq: Population-Sclae COVID-19 Diagnostics Using Combinatorial Barcoding. bioRxiv 2020.04.06.025635.
The Rockefeller Foundation. National Covid-19 Testing Action Plan Pragmatic steps to reopen our workplaces and our communities. 2020.
Cahill TJ, Cravatt B, Goldman LR, Iwasaki A, Kemp RS, Lin MZ et al. Scientists to Stop COVID-19. OR Rob Copeland, Wall Street Journal (2020) The Secret Group of Scientists and Billionaires Pushing a Manhattan Project for Covid-19. April 27
-Jeff SoRelle, MD is a Chief Resident of Pathology at the University of Texas Southwestern Medical Center in Dallas, TX. His clinical research interests include understanding how the lab intersects with transgender healthcare and improving genetic variant interpretation.
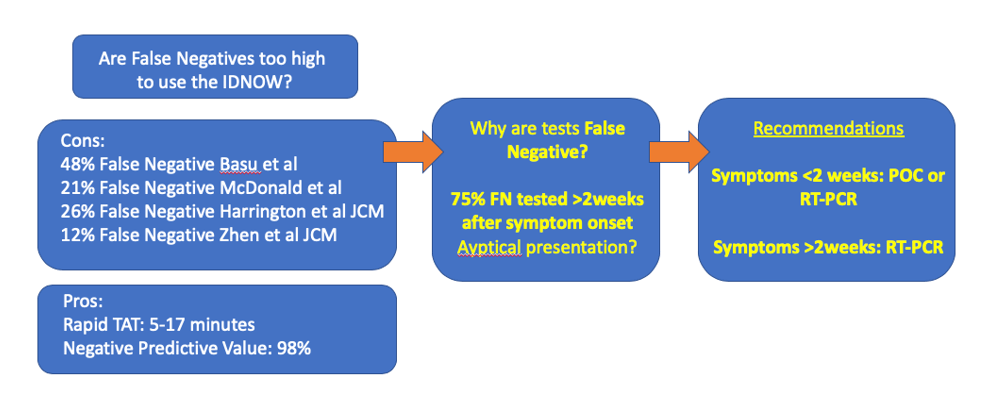
I left for vacation at the beginning of June thinking “once I get back, all of this COVID stuff will be quieted down.” …Well that wasn’t quite the case and testing for novel Coronavirus has continued to be very important. In fact, this last weekend I was tested by occupational health. It came back negative, but I’m am very enthusiastic to get alternative specimen types validated; those Nasopharyngeal swabs are quite…uncomfortable. Luckily, my test was processed at our institution which gets results back in 24-48 hours. However, with the resurgence around the country, turnaround times are backing up to 7-8 days. One solution has been the widely used IDNOW point of care platform. However, there has been significant concern over false negatives produced by this platform. One reason the sensitivity is different is because this platform performs isothermal amplification of nucleic acid. This method amplifies RNA at a stable temperature instead of cycling the temperature as in real-time PCR.
Colleagues at my institution reflexed any negative IDNOW samples to the m2000 Real-Time PCR assay for SARS-CoV-2 for one month. Within that time, over 500 samples were tested and the IDNOW was found to have missed 21% of positive cases (prevalence rate of 5%)2. One the positive side, it had a 98% negative predictive value, which helped rule out COVID19 infection. However, as prevalence rates are increasing, a high negative predictive value isn’t as important as sensitivity.
One study drew much attention when it claimed the IDNOW had a sensitivity of 52% in a New York City academic institution (Basu)4. However, this seems to be an outlier compared to other studies of this platform: one large multi-center study found positive percent agreement (equivalent of sensitivity when a gold standard test hasn’t been established) of 74%1. The highest PPA of 88%3 for the IDNOW was found in a study that indicated it can be completed in 17 minutes, whereas another quick instrument (but not point of care instrument: Xpert Xpress, 45min) had a PPA of 98%2.
Myself and other colleagues looked more closely at the clinical characteristics of false negative test results on the IDNOW. Overall, we found 82% PPA, and 8 patients with false negative tests. Interestingly, a majority of these patients were tested over 2 weeks after their initial onset of symptoms. The virus is known to be at its highest levels at the beginning of symptom onset. So the test may not be limited, but it should be used in the correct clinical context (< 2weeks from symptom onset). After that time, other RT-PCR based tests are more appropriate.
As clinical laboratorians, we often hear: “the right test for the right patient at the right time.” Now with so many platforms available for use in different contexts, we should help guide clinicians to Choose Wisely.
References
Harrington A et al. Comparison of Abbott ID Now and Abbott m2000 methods for the detection of SARS-CoV-2 from nasopharyngeal and nasal swabs from symptomatic patients. JCM 2020. PMID: PMID: 32327448
McDonald et al. Diagnostic Performance of a Rapid Point of Care Test for SARS-CoV-2 in an Urban ED Setting. Academ. Emerg. Med. 2020. PMID: 32492760
Zhen W et al. Clinical Evaluation of Three Sample-To-Answer Platforms for the Detection of SARS-CoV-2. JCM 2020. PMID: 32332061
Basu A et al. Performance of the rapid Nucleic Acid Amplification by Abbott ID NOW COVID-19 in nasopharyngeal swabs transported in viral media and dry nasal swabs, in a New York City academic institution. BioRxiv 2020.
-Jeff SoRelle, MD is a Chief Resident of Pathology at the University of Texas Southwestern Medical Center in Dallas, TX. His clinical research interests include understanding how the lab intersects with transgender healthcare and improving genetic variant interpretation.
This month we will finish the discussion the common barriers to biomarker testing for cancer patients in the community. Lengthy complex reports is a relatively straightforward barrier to address, so I will pair it with the lack of education on guidelines barrier to complete this blog series on barriers to biomarker testing.
As you may recall, these are the top 10 barriers that I’ve seen to biomarker testing in the community:
High cost of testing.
Long turnaround time for results.
Limited tissue quantity.
Preanalytical issues with tissue.
Low biomarker testing rates.
Lack of standardization in biomarker testing.
Siloed disciplines.
Low reimbursement.
Lengthy complex reports.
Lack of education on guidelines.
Lengthy Complex Reports
Laboratory issued reports are typically developed by the lab and are often written in a manner that is easy to understand for other laboratorians. I’m guilty of writing long interpretive comments that are attached to every molecular diagnostics results. I would get irritated when the physician would call and ask questions that in my mind were clearly addressed in the interpretive comment. I thought the issue was they were not reading the comments (and this could be true). I now understand that the issue is that the comments were not written for the end-user.
When insourcing NGS I was fortunate enough to get feedback from the multidisciplinary team in the Molecular Steering Committee. One of the complaints that I heard loudly locally, that also resonated in the community, was the reports for NGS were way too long and they didn’t find value in half of the information that was in the report. When were shopping for the right cloud-based reporting software, I kept the feedback in mind from the oncologists. I was actually able to get proto-type reports from 3 different companies and provide them to the oncologists for them to score and provide feedback on the layout. This was invaluable in developing a report that worked well for the treating physician and not the laboratory.
Some of the feedback they gave that made a direct impact into the report we created was: bold the patient’s name so they can easily find it, use patterns as well as color-coding for drug resistance/sensitivity in case the document is faxed, and tell them everything they need to know to make treatment decisions on page one. These are things that were not intuitive to me. Having end-user feedback helped us generate a more useable report and enlightened me that the report needs to be written to an oncology audience.
Lack of education on guidelines
I’ve had the opportunity to do a great deal of educating around biomarker testing in the community. Physicians and nurses in the community want to provide guideline-driven care. Often when we are educating on changes to guidelines, it’s the first time the providers have heard of the change. NCCN for lung cancer alone had at least 7 updates in 2019. It’s amazing that the guidelines are able to keep up with the ever changing science and drug approvals; however it’s incredibly difficult to keep track of the changes.
In large institutions we are fortunate enough to have specialized physicians that help keep the rest of us informed of changes in their area of expertise. Community physicians typically see and treat all types of cancers and don’t always have the network of specialists to keep them informed of changes for every cancer type. Many of them also do not have the time to attend conferences due to heavy workload.
In order for the community physician to be informed of all of the changes to guidelines for every tumor type, we need to make sure the information is provided in a variety of methods. The information needs to be easily accessible. I have found that educational programs work well when brought to the community rather than trying to get the community to come to them. Pharmaceutical and diagnostic companies and even reference laboratories now have teams of individuals in roles that are intended to educate and not sell. They can provide in office education, facilitate webinars, lunch and learns, and dinner programs. If there is a champion for biomarker testing within the facility, you can develop your own educational program to be delivery locally at grand rounds. We discuss changes to guidelines within our Molecular Steering Committee. I’ve also talked to institutions where this education is given during tumor boards.
I don’t think there is a bad forum for education. Some physicians may prefer getting guideline updates from twitter; others will be more comfortable with a discussion with an expert, regardless of the medium it is important that we help facilitate education of guidelines in order to increase biomarker testing rates in the community.
-Tabetha Sundin, PhD, HCLD (ABB), MB (ASCP)CM, has over 10 years of laboratory experience in clinical molecular diagnostics including oncology, genetics, and infectious diseases. She is the Scientific Director of Molecular Diagnostics and Serology at Sentara Healthcare. Dr. Sundin holds appointments as Adjunct Associate Professor at Old Dominion University and Assistant Professor at Eastern Virginia Medical School and is involved with numerous efforts to support the molecular diagnostics field.
Much has changed quickly with SARS-CoV-2 virus (COVID-19) testing. Several commercial options are now available. Labs have less problems getting control material (positive samples are no longer in short supply). And labs that opted to bring on testing are now running multiple versions of COVID-19 molecular tests with a combination of high speed platforms or high throughput. Rapid cartridge tests are used for clearing people from the ED/ removing contact isolation on inpatients while the high throughput assays are used for routine screening.
However, several bottlenecks still exist. There are shortages of nucleic acid extraction kits, collection swabs and viral transport media. Fortunately, some recent studies have demonstrated preliminary evidence for using alternative sample types, collection methods, and storage conditions.
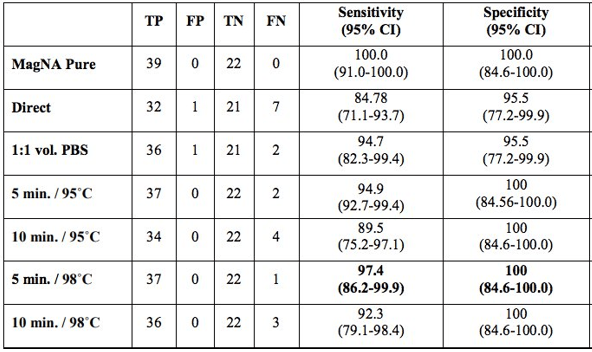
One of the first tenets of molecular diagnostics is isolation and purification of nucleic acid. Therefore, it was surprising to see a report on an extraction-free COVID-19 protocol from Vermont (Bruce EA et al.). This study initially analyzed two patient samples and showed drops in sensitivity of ~4Ct cycles. While this would not be suitable for low level detection, many viral samples have high levels of virus that still would permit detection. The team went on to test this method on 150 positive specimens from the University of Washington and found 92% sensitivity with 35% sensitivity at the low viral load range (Ct value> 30). This was improved with a brief heat inactivation step (Table 1). This was similarly seen in a study from Denmark, where brief heat inactivation of extraction-free methods (Direct) had 97% specificity in 87 specimens (Table 2).
Table 1. Vermont study comparing sensitivity of direct RT-PCR (no extraction step) with the validated results of 150 specimens coming from the University of Washington.Table 2. Denmark study found extraction-free protocols (Direct) were comparable to extracted RNA (MagNA Pure extraction method) detection in 87 specimens.
Some similar studies out of Chile also showed extraction-free protocols on a larger number of specimens, and they reported a loss in sensitivity varying from 1-7 Ct cycles depending on the primers used.
Figure 1. P1 and P2 are patient 1 and 2. NSS indicates a nasal swab sample where RNA was extracted. RNA indicates a sample with no RNA extraction.
As this novel Coronavirus has an RNA-based genome, RNA is the target of molecular tests. As RNA is susceptible to degradation, there have been concerns over sample storage. Should it be refrigerated? Frozen? How do multiple freeze-thaw cycles impact specimen stability? Are there viable alternatives to viral transport media? One preliminary study explored these questions very nicely. They took X multiple sample types (NP, BAL, saline storage media) and stored them at 20C, 4C, -20, and -70 for multiple days up to 1 week and then analyzed the level of virus detected. In each case, the loss in sensitivity was minimal (<2 Ct cycles from day 0 to day 7) at room temperature with comparable results at lower temperatures (Table 3).
Table 3. Stability of SARS-CoV-2 RNA detected by the Quest EUA rRT-PCR. VCM- viral culture media; UTM-R Copan’s transport medium; M4-microtest media; BAL- bronchoalveolar lavage.
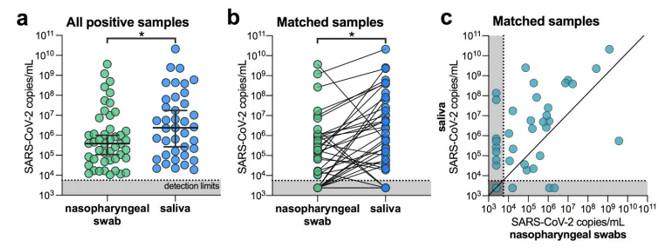
Lastly, alternative sample types such as saliva will help break the bottleneck in swabs and viral transport media. I was surprised to hear about this being a suitable alternative. Having worked with saliva for DNA analysis, I know it can be contaminated, of variable quantity, includes digestive enzymes and is viscous (slimy). These are not characteristics a lab would look for in a specimen type being used for high-throughput testing where several sample failures could occur. But these researchers from Yale showed measurable levels of SARS-CoV-2 that facilitated even higher sensitivity than nasopharyngeal swabs (Wylie AL et al).
Figure 2. SARS-CoV-2 titers are higher in the saliva than nasopharyngeal swabs from hospital inpatients. (a) All positive nasopharyngeal swabs (n = 46) and saliva samples (n = 39) were compared by a Mann-Whitney test (p < 0.05). Bars represent the median and 95% CI. Our assay detection limits for SARS-CoV-2 using the US CDC “N1” assay is at cycle threshold 38, which corresponds to 5,610 virus copies/mL of sample (shown as dotted line and grey area). (b) Patient matched samples (n = 38), represented by the connecting lines, were compared by a Wilcoxon test (p < 0.05). (c) Patient matched samples (n = 38) are also represented on a scatter plot.
With a much-needed increase in testing for this country, optimizations need to be implemented to improve efficiency. These steps alone will not be enough, but if we can have extraction-free testing of saliva collected at home, this would provide a substantial benefit to bringing easy testing to everyone.
UPDATE: Since this was written, the first FDA EUA was authorized for an at-home saliva collection kit for use at the Rutger’s clinical genomics lab (https://www.fda.gov/media/137773/download).
References
Please note: many of these references were on pre-print servers and have not been peer-reviewed.
Rogers AA, Baumann RE, Borillo GA, et al. Evaluation of Transport Media and Specimen Transport Conditions for the Detection of SARS-CoV-2 2 Using Real Time Reverse Transcription PCR. JCM 2020.
-Jeff SoRelle, MD is a Chief Resident of Pathology at the University of Texas Southwestern Medical Center in Dallas, TX. His clinical research interests include understanding how the lab intersects with transgender healthcare and improving genetic variant interpretation.
I hope everyone is staying safe and healthy during these unfortunate times. My last post was in relation to being a new MLS grad and beginning my career as a molecular technologist at Northwestern University’s Transplant Lab. Time definitely flies by!
Today I’m going to provide a basic introduction on an assay I’ve recently been trained on called Short Tandem Repeat (STR). If you were to take a glance at your genome, it would be littered with many repeating sequences. While there are many different classifications of repeating sequences, STRs are a type of tandem repeating sequence where each repeat is approximately 2 to 7 nucleotides in length.1,2,3 STR is well-known in forensic science to help identify a suspect at a crime scene when different sources of DNA are present. Yet, its applications are many – from cell line confirmation, paternal testing, and all the way to chimerism analysis!4
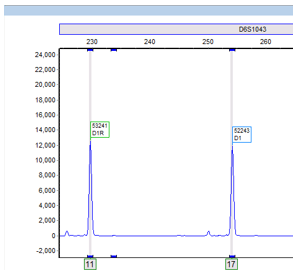
Image 1. Electropherogram depicting two different alleles (11 and 17) within 1 locus (D6S1043). Allele 11 has 11 repeats and allele 17 has 17 repeats.
STRs are polymorphic, one useful characteristic among many, which make its utilization in identifying the source of DNA particularly advantageous. An STR allele is defined by the number of times the repeating sequence, defined above, repeats. (Image 1).1 Individuals are either heterozygous or homozygous at each locus. As the number of STR loci being evaluated increases, the statistical power of discrimination increases and the likelihood of another individual having the same profile becomes increasingly unlikely and detecting small differences increases.3,4 In our lab, we evaluate a total of 21 different loci!
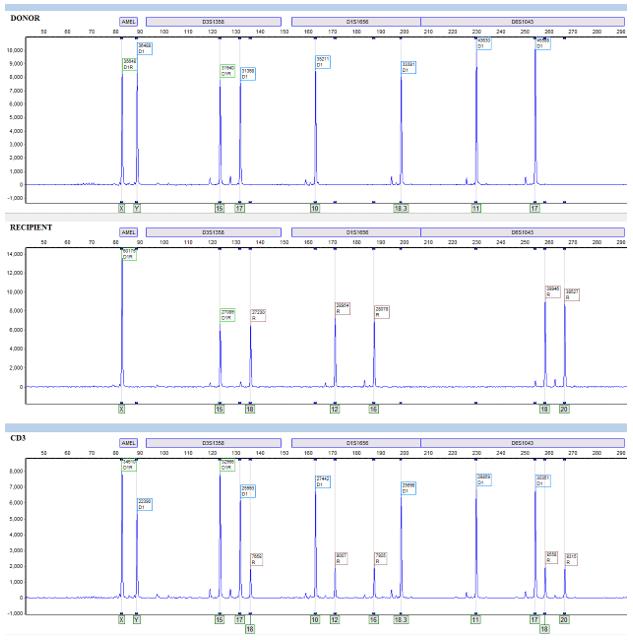
Additionally, in our HLA lab we utilize STR to monitor chimerism status in patients who have undergone an allogeneic stem cell transplant. Before their transplant, patients are matched to a donor through their HLA system (different from STR). Once an HLA match is confirmed, we utilize the patient’s pre-transplant and the donor’s sample to generate STR informative alleles. Informative alleles are alleles that are present only in the recipient and not the donor. These alleles are important because stem cell transplants replace the recipient’s marrow and the detection of recipient DNA in post-transplant samples is crucial to identifying rejection or relapse of their disease. Additionally, loci that contain informative alleles are defined as informative loci, these are the loci then used to identify the percentage chimerism (Image 2).
Image 2. Donor and recipient (pre-transplant) are represented in the first two electropherograms. The green “D1R” tags represent shared alleles, the blue “D1” tags represent donor specific alleles, and the brown “R” tags represent recipient specific alleles. In the first locus, AMEL, there are no informative alleles and therefore it is not an informative locus. In the next locus, D3S1358, there is one shared allele, 15, one donor allele, 17, and one recipient informative allele, 18. Informative loci have recipient informative alleles and can detect the presence of recipient DNA in a sample – in this case all of the following loci after AMEL are informative loci. CD3 (post-transplant) is represented on the third electropherogram. In this example, it is clear that the patient is having some sort of graft failure or reoccurrence of their disease because their own cells, instead of just the donor, are present.
When recipient cells begin to re-emerge, we can detect the relative allele peaks and assign them to the recipient or donor by referring to the informative alleles. Allele peaks are then utilized through equations in our software that measure the area under these defined peaks and then compute a donor percentage chimerism. Once that informative report is created, we can compare any proceeding post-transplant sample to determine the patient’s chimerism status (Image 2).
Interesting enough, we don’t simply isolate the DNA from the post-sample buffy like we would with other samples. Rather, we separate each post sample into a total of three sub-samples. The first is simply the patient’s peripheral blood – nothing fancy. The other two are isolated from the rest of the peripheral blood that was not used into two separate cell lineages – lymphocytes (CD3+) and myelocytes (CD33+). This process is extremely labor-intensive, being able to process a set of up to 8-12 patients at a time and each set taking up to 4-5 hours.
The process begins by aliquoting peripheral blood for DNA isolation (sample 1) and taking the remainder and layering it over a lymphocyte separation medium (LSM). Then harvesting the lymphocyte/white cell layer from the spun down LSM. We then go on to add CD3 and CD33 antibody selection cocktails and magnetic beads. Then, we do a series of washes with magnets and eventually end up with our purified CD3 and CD33 cell populations. Their purity is determined through flow cytometry, an important component to confirm that our leukocyte subsets aren’t contaminated with other leukocyte populations – as contamination would defeat the purpose of analyzing different lineages.
Finally, we take the isolated DNA from the three sub-samples and amplify it with specific primers and fluorescent tags through PCR. Then the samples are loaded onto a capillary electrophoresis instrument. This instrument will detect each fragment length, defined by the primers and the repeats within, and be able to identify these fragments through size, fluorescent tags, lasers, and detectors. The instrument will then generate data that we can take to our analyzing platform, which is ChimerMarker. Through this we can analyze the data and generate our clinical reports.
Though extremely time intensive, lineage-specific chimerism is critical in stem cell transplant because it is more informative and sensitive than total leukocyte analysis – being several magnitudes more sensitive than analyzing just peripheral blood alone. It permits early detection of small chimeric cell populations that otherwise may go undetected, as one subset in the peripheral blood may “mask” another subset that has increasing percent recipient cells. Diagnosing these small cell chimeric cell populations as early as possible is critical for therapeutic interventions and reductions in graft rejections.2,5,6
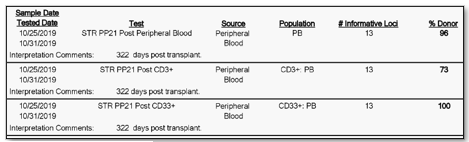
Furthermore, not only is their detection important, but through our analysis we can calculate the percentage of donor cells and recipient cells. We oftentimes report out the donor percentage (%) chimerism. For example, a patient at 322 days post-transplant could have a donor chimerism of 96% in their peripheral blood, 100% in their CD33 lineage, and 73% in their CD3 lineage. Then, at day 364 post-transplant they may then be at 100% in their peripheral blood, 100% in their CD33 lineage, and 92 percent in their CD3 lineage. Two things to notice in this example is that the percentages are changing (increasing in donor chimerism in this case) and that the peripheral blood expressed 100% chimerism in the second sample at 364 days, but when we look specifically at the CD3 sub-population at 364 days there was still 8% of recipient cells present (Image 3 & 4).
Image 3. Samples at 322 days post-transplant. Peripheral blood reports 96% donor chimerism. CD3 and CD33, purified from peripheral blood, reports 73% and 100% donor chimerism, respectively.Image 4. Samples at 364 days post-transplant, same patient as in Image 3 above. Peripheral blood reports 100% donor chimerism. CD3 and CD33, purified from peripheral blood, reports 92% and 100% donor chimerism, respectively.
Some studies have focused not only on the trends in percentages changing, but also in their relative percentage constellation. For example, one study found that increased recipient CD3 cells had an increased predictive factor of graft rejection. It was also found that further sub-leukocyte populations increased this predictive power.5 Even more, there have been some studies that have looked at chimerism and its usefulness in predicting graft versus host disease (GvHD). This disease is defined by donor leukocytes attacking the leukocytes and tissues of the recipient. Through these and other findings, the potential and applicability of chimerism monitoring is extremely crucial to patient care during their transplant progression.2,5,6,7
While engraftment is a very dynamic process, varying from individuals and disease-types, engraftment monitoring is one way to monitor and ultimately influence therapeutic approaches.2,5,6 I am proud to be able to contribute to the wonderful team here at Northwestern University and I strive to learn more about the process – both clinical and in the lab. In future articles, I hope to go into more detail about the process and other assays that we perform.
Thanks for reading! Until next time! Stay well and safe during these uncertain times!
Kristt, D., Stein, J., Yaniv, I., & Klein, T. (2007). Assessing quantitative chimerism longitudinally: technical considerations, clinical applications and routine feasibility. Bone Marrow Transplantation, 39(5), 255–268. doi: 10.1038/sj.bmt.1705576
Clark, J.R., Scott, S.D., Jack, A.L., Lee, H., Mason, J., Carter, G.I., Pearce, L., Jackson, T., Clouston, H., Sproul, A., Keen, L., Molloy, K., Folarin, N., Whitby, L., Snowden, J.A., Reilly, J.T. and Barnett, D. (2015), Monitoring of chimerism following allogeneic haematopoietic stem cell transplantation (HSCT): Technical recommendations for the use of Short Tandem Repeat (STR) based techniques, on behalf of the United Kingdom National External Quality Assessment Service for Leucocyte Immunophenotyping Chimerism Working Group. Br J Haematol, 168: 26-37. doi:10.1111/bjh.13073
Breuer, S., Preuner, S., Fritsch, G., Daxberger, H., Koenig, M., Poetschger, U., … Matthes-Martin, S. (2011). Early recipient chimerism testing in the T- and NK-cell lineages for risk assessment of graft rejection in pediatric patients undergoing allogeneic stem cell transplantation. Leukemia, 26(3), 509–519. doi: 10.1038/leu.2011.244
Buckingham, L. (2012). Molecular diagnostics: fundamentals, methods, and clinical applications. Philadelphia: F.A. Davis Company.
Rupa-Matysek, J., Lewandowski, K., Nowak, W., Sawiński, K., Gil, L., & Komarnicki, M. (2011). Correlation Between the Kinetics of CD3 Chimerism and the Incidence of Graft-Versus-Host Disease in Patients Undergoing Allogeneic Hematopoietic Stem Cell Transplantation. Transplantation Proceedings, 43(5), 1915–1923. doi: 10.1016/j.transproceed.2011.02.011
-Ben Dahlstrom is a recent graduate of the NorthShore University HealthSystem MLS program. He currently works as a molecular technologist for Northwestern University in their transplant lab, performing HLA typing on bone marrow and solid organ transplants. His interests include microbiology, molecular, immunology, and blood bank.
This month we will continue discussing the common barriers to biomarker testing for cancer patients in the community.
As you may recall, these are the top 10 barriers that I’ve seen to biomarker testing in the community:
High cost of testing.
Long turnaround time for results.
Limited tissue quantity.
Preanalytical issues with tissue.
Low biomarker testing rates.
Lack of standardization in biomarker testing.
Siloed disciplines.
Low reimbursement.
Lengthy complex reports.
Lack of education on guidelines.
When I go into the community and discuss barriers to biomarker testing while everyone can relate to 1-2 barriers, those barriers are typically not the same at every hospital. However, reimbursement is almost always presented as a barrier to biomarker testing. The reimbursement process may be confusing and there have been recent changes. If everything is not submitted properly, testing may not be covered. Let me start by saying I have no magic bullet to fix the problems with molecular pathology billing and I’m not the expert on billing. I have had to navigate the reimbursement process and can share my experiences.
Let’s start with Medicare as they represent a payer all of us have to work with and we frequently see other insurers make coverage decisions based on Medicare rates. The Medicare coverage for single gene testing has historically covered the testing, albeit maybe not at a rate we consider acceptable. In 2018 Medicare issued a national coverage determination (NCD) for NGS if the patient has stage III or IV cancer and the NGS assay has an FDA-approved or cleared indication for use in that patient’s cancer and results are provided to the treating physician for management of the patient using a report template to specify treatment options (1). This means if you use a reference laboratory that has an assay that is approved as a companion diagnostic for a drug that is approved in the tumor type you are testing, the test could be covered. For the test to be covered the correct CPT code from the AMA would need to be applied, an ICD-10 qualifying code to meet medical necessity, and if your state is covered by the MolDX program you would also need to provide a Z-code that is specific for the test. Confused yet?
There is also a Medicare 14-day rule (formally called Date of Service Regulation 42 C.F.R. §414.510). This rule requires the performing lab to bill the hospital for certain tests that are ordered less than 14 days after an inpatient or outpatient discharge. There was a change as of January 1, 2018 that allows labs to bill for certain molecular pathology tests if the patient was admitted as an outpatient (think biopsy performed in hospital but patient was not admitted as an inpatient). This does not negate the 14 day rule, but it gives us some exceptions so that we may bill for molecular pathology testing ordered after the patient was discharged. This rule also mandates that the performing lab is the billing lab.
For payers that are not Medicare, it is helpful to have a conversation with the medical director or a customer service representative to get information on how to get your test covered. We have presented to the medical directors for private payers. While we did cover the scientific merit of our testing, we also had to go over financials for the payer. It was helpful to speak their language and provide clear information on the financial benefit to NGS over single gene testing.
Many of the reference laboratories will handle the billing for you if your hospital contract with them is written that way. This would allow those of us that are not billing experts to ensure all of the coding is applied properly. Of course you would still need to supply the information to the reference laboratory. These labs also offer low out of pocket costs to the patient. If you are insourcing testing, I would recommend having a molecular billing consultant. There are consultants available that allow you to submit questions and pay per question. This has come in handy for my organization.
Lastly, I urge you to join and get involved with organizations that represent the laboratory community such as CAP, AMP, ASCP, etc. These organizations help address policy change to ensure molecular testing is reimbursed in a fair manner. Molecular pathology results have value for the patient and cost money to be performed. We should expect fair payment for the service rendered.
Reference
National Coverage Analysis (NCA) for Next Generation Sequencing (NGS) for Medicare Beneficiaries with Advanced Cancer (CAG-00450N). 1/21/19
-Tabetha Sundin, PhD, HCLD (ABB), MB (ASCP)CM, has over 10 years of laboratory experience in clinical molecular diagnostics including oncology, genetics, and infectious diseases. She is the Scientific Director of Molecular Diagnostics and Serology at Sentara Healthcare. Dr. Sundin holds appointments as Adjunct Associate Professor at Old Dominion University and Assistant Professor at Eastern Virginia Medical School and is involved with numerous efforts to support the molecular diagnostics field.
Since I last wrote about some testing options available for COVID-19 testing just 1 month ago, many things have changed in the regulatory requirements, and the companies offering testing options. With that in mind along with the fact that things will likely continue to changes, I’ll write to address current and future challenges facing COVID-19 laboratory testing.
What control material can be used?
How can I make specimens safe?
Supply chain issues and solutions.
False Negative results of COVID-19 tests: what to tell clinicians.
Serology Tests: Future Testing and Challenges
Control Material
As the FDA said contrived specimens could be used, that means that RNA can be spiked into a clinical matrix for extraction. While this began with a requirement for genomic RNA, it has been loosened to include plasmid DNA. However, I would caution against using plasmid DNA, because when it is amplified, it can easily cause contamination and unlike RNA, DNA can persist in the environment for a long time. I once hear a story about a lab director who thought they were very careful, but in pipetting, they contaminated the lab in 3 days and it had to be cleaned up for 4 weeks.
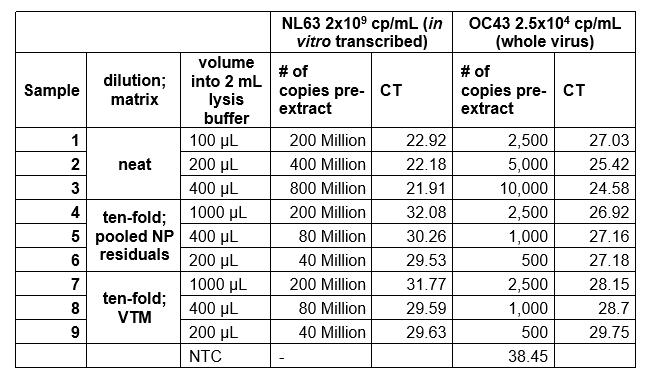
We had some issues using in vitro transcribed RNA (of just the N-gene) and genomic RNA, because the recovery was very low. We found out that intact viral particles were better in optimization experiments using control endemic SARS strains (Zeptometrix controls, Table 1). The free RNA Ct values fell sharply (over 1000-fold) when added to Nasopharyngeal (NP) matrix or Viral Transport Media (VTM). However, much lower levels of the encapsulated viral control had consistent levels of amplification.
Table 1. Amplification of free viral RNA vs. viral particles when added to matrix
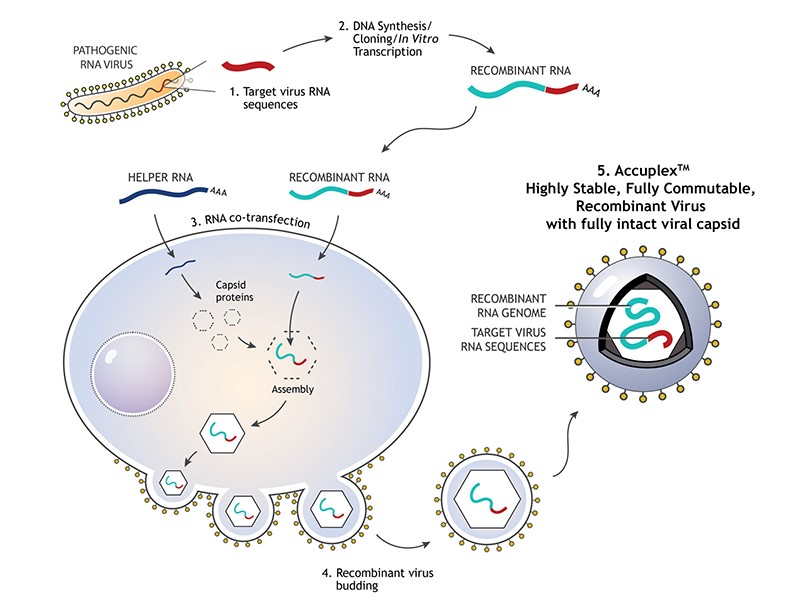
Therefore, we used a synthetically encapsulated SARS-CoV-2 RNA sample called Accuplex (SeraCare, Figure 1), which gave good recovery and a limit of detection down to 260 copies/ mL (5 copies/ reaction). Alternative similar material that we have not evaluated include: COVID-19 RNA synthesized inside inactivated E coli (Zeptometrix) and Armored RNA (Asuragen).
Figure 1. Schematic of how a synthetically created viral particle occurs
Lab Safety of Specimens
A safety recommendation of the FDA was to perform extraction of samples in a Biosafety level 2 hood. However, high-throughput extraction can’t be easily done this way. For us, we had to prepare samples in the hood then take them to the stand-alone closed system extractor. Our Micro fellow had the creative idea to do “Off-Board” lysis, which would inactivate the virus in the hood before walking it over. We later found that combining lysis with NP matrix before spiking RNA stabilized the RNA for accurate measurement. We were able to find an LOD of 14 copies/ reaction this way.
Some labs have proposed using heat inactivation (~30 minutes at 50-60C) of virus as a safety measure, but the published literature available on how that affects sensitivity is lacking currently.
Supply Chain issues
You have surely heard about all of the new companies that have come out with new testing platforms and assays for COVID-19 testing by now. However, the downside is that unless you already have their instrument, you likely won’t be able to get reagents in time to perform the assay. Even if you do have an instrument, limited resources are necessitating allocation based on high risk areas, so you are likely to receive fewer kits than you would like. Also, the reference labs are still ramping up capacity and are returning results back with long turnaround times currently (~ 1 week). This supports the strategy to bring the testing in-house, so that you can get results back quickly and have control over at least your labs reagent supply. If you have the instrumentation of another FDA approved EUA, you can start performing testing if you follow that protocol exactly- the CDC is the most widely used.
False Negative Tests of COVID-19
This is hard to assess when only one lab testing modality (PCR) is available, but clinicians report negative results in a patient with classic symptoms and a contact history with a COVID-19+ person. Given the impressive analytic sensitivity of the test (generally 1 copy of RNA in 1mL of sample), the likely explanation is that there is a specimen issue. Proper NP sampling requires sticking the swab to the very back of the nasal cavity. Furthermore, this virus may reside more in the lower respiratory track (lungs) and simply not be present in the area sampled. This is why repeat sampling could be helpful. However, the outcome should be the same whether or not you have a negative test: if you have symptoms you should self-isolate unless you require emergent care due to shortness of breath or other symptoms that can’t be managed at home. The lab is familiar with these pre-analytic limitations that can arise, but it is helpful to explain this to clinicians.
Serology Tests: Future Testing and Challenges
Serology can be very helpful as a separate method from qPCR to determine if someone has been infected with SARS-CoV-2. Notice, that is in the past tense. A small (n=9) pre-print study from Nature indicates serologic conversion starts around day 8 or 10 after symptom onset, which often is not in a clinically helpful timeframe. However, these tests are cheaper and easier to perform, so could be useful for epidemiological purposes to determine who has been infected with COVID-19. Early genetic data indicates that the mutation rate is slow (4x slower mutation rate compared to seasonal flu) as one would suspect for RNA-based viruses, so the virus shouldn’t change enough to cause re-infection in someone with sufficient antibody levels.
Figure 2. Scheme of the first COVID-19 antibody test to receive FDA Emergency Use Authorization.
However, several challenges in interpreting these antibody tests include:
Some conflicting data as to how quickly IgM develops relative to the onset of symptoms
What is the time-line for IgG production?
Ruling out cross reactivity with other strains of Coronavirus that cause upper respiratory infections.
Mitchell S, George K et al. Verification procedure for commercial tests with Emergency Use Authorization for the detection of SARS-CoV-2 RNA. American Society of Microbiology
-Jeff SoRelle, MD is a Chief Resident of Pathology at the University of Texas Southwestern Medical Center in Dallas, TX. His clinical research interests include understanding how the lab intersects with transgender healthcare and improving genetic variant interpretation.
This month we will continue discussing the common barriers to biomarker testing for cancer patients in the community.
As you may recall, these are the top 10 barriers that I’ve seen to biomarker testing in the community:
High cost of testing.
Long turnaround time for results.
Limited tissue quantity.
Preanalytical issues with tissue.
Low biomarker testing rates.
Lack of standardization in biomarker testing.
Siloed disciplines.
Low reimbursement.
Lengthy complex reports.
Lack of education on guidelines.
Despite being unique hurdles, a few of these barriers can be addressed together. If you are able to standardize biomarker testing despite the barriers that come with being in siloed disciplines, biomarker testing rates will go up. Sounds easy right! I am a firm believer in the multidisciplinary approach to precision medicine, because I have seen it work in my institution. I have also spoken with organizations where there is no collaboration among the multidisciplinary team (MDT) and observed what happens when the team is not working together. In these cases biomarker testing is often not being performed according to guideline and relationships between the pathology and oncology are strained.
In my organization we have a lot of complexity: 12 hospitals, inhouse and external reference labs, our own private payer, and pathology and oncology groups that are not related to the organization or each other. Everyone wants to do what’s right for the patient, unfortunately if everyone is not working together to help the patient, we tend to get in each other’s way. We found that our oncologists were not getting results back on biomarker tests in reasonable amount of time to make educated treatment decisions. The oncologist chose when to order testing, which biomarker to test, and the performing lab. This resulted in a great deal of variance in the care provided by each physician. It also added complexity in the pathology laboratory. We had to have shipping containers, portals, collection and specimen requirements that were different for every reference laboratory that the oncologists used. This delayed turnaround time even more as we navigated through the nonstandard process for biomarker testing. As you can imagine tensions were high between pathology and oncology.
Our organization began following the high performance team model some years ago. With this model we have a “team of teams” that can effect change rapidly despite a complex organizational structure (1). Every stakeholder is represented in the meeting, without every stakeholder having to attend the meeting. So if you have a team of oncologists that already trust their colleague they are typically comfortable allowing one oncologist to represent their best interest in the committee. We now have a vast structure of committees built on the principle of extending trust from one group into another group with stakeholder representation to build relationships between teams.
One of these committees is a Molecular Steering Committee. I co-chair this committee along with an oncologist. It is attended by radiologists, pathologists, oncologists, administrators and even the medical director from our payer. Every stakeholder and geographic region is represented. In this committee we discuss how to standardize biomarker testing by tumor type. Although our committee is distinct from a molecular tumor board where you can discuss molecular results for cases, any forum where standardizing the biomarker process can be addressed with a multidisciplinary team is the right forum. We have built relationships between the stakeholders involved in biomarker testing and help keep each other educated on changes to guidelines across tumor types.
This committee has allowed us to develop pathology-driven reflexes for testing in specific scenarios. Not all biomarker testing can or should be done at the time of diagnosis. However, some tumor types such as NSCLC adenocarcinoma where the tissue is limited and turnaround time is urgent, it makes a lot of sense to perform the testing as soon as we know the patient has this disease. In these cases the pathologist orders NGS and PD-L1 testing when they determine the diagnosis. This drastically cuts down on the turnaround time (2 weeks vs 6 weeks) and has the added benefit of ensuring all patients with this diagnosis get the standardized biomarker testing that they deserve.
Having a multidisciplinary forum to discuss biomarker testing by tumor type, including which tumor types, what stage, who’s ordering (pathology vs oncology), which test, and where it is performed is necessary to bridge the gap between siloes. In some institutions this can be done without a formal committee, a phone call between oncology and pathology may suffice. The most important thing you can do to improve your biomarker testing rates and increase standardization is to communicate across silos or disciplines to ensure everyone is in alignment on how to determine patients’ biomarkers status.
Reference
McChrystal, T. C. D. S. C. F. S. A. (2015). Team of Teams: New Rules of Engagement for a Complex World.
-Tabetha Sundin, PhD, HCLD (ABB), MB (ASCP)CM, has over 10 years of laboratory experience in clinical molecular diagnostics including oncology, genetics, and infectious diseases. She is the Scientific Director of Molecular Diagnostics and Serology at Sentara Healthcare. Dr. Sundin holds appointments as Adjunct Associate Professor at Old Dominion University and Assistant Professor at Eastern Virginia Medical School and is involved with numerous efforts to support the molecular diagnostics field.
The FDA is now democratizing the testing of the novel coronavirus: SARS-CoV-2 (the virus which causes the COVID-19 disease syndrome—I will call it COVID-19 from here on as that is the colloquial name most people know) by allowing high complexity testing labs across the United States. This move will permit more labs to test for COVID-19. A previous post by contributor Constantine Kanakis describes the biology of the virus, so I will not repeat that material. Instead, I will focus on some considerations in validating a Lab Developed Test (LDT) COVID-19 molecular assay.
The president of AACC, Carmen Wiley, said there are 11,000 high complexity testing labs in the US, which could qualify for performing this testing. However, not all of these labs have molecular and virology expertise, so others have placed the number of labs with qualified staff and instrumentation at 400.
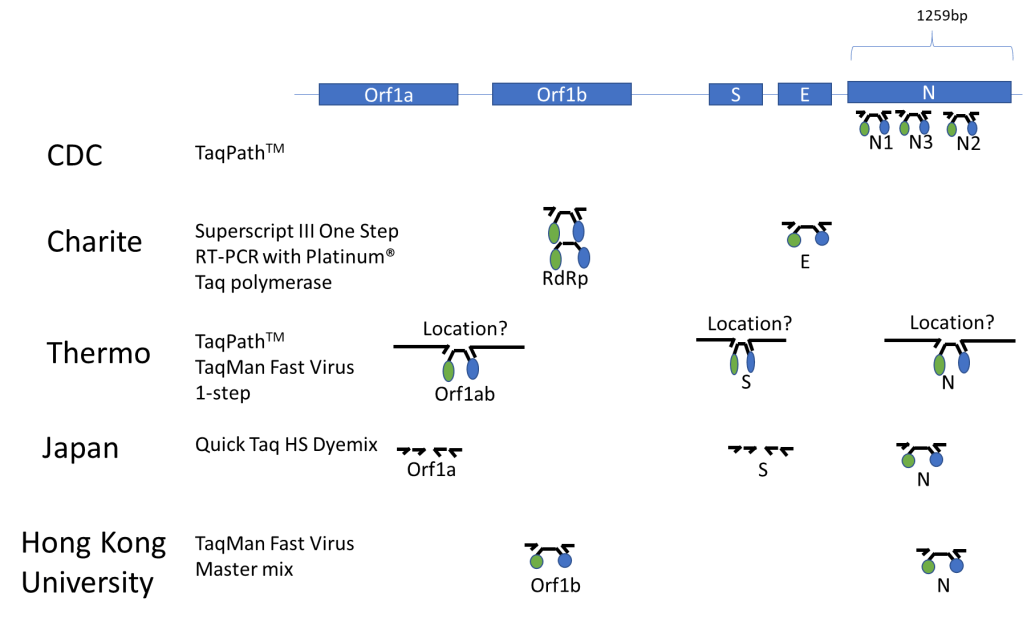
Published Assays and Targets: As an overview, the figure below (Figure 1) summarizes some published COVID-19 assays. As you can see, the major strategy involves using the TaqMan probe strategy where a short probe is degraded by Taq polymerase releasing a fluorescent molecule (green ball) from a quencher molecule (blue ball). The TaqMan approach allows for quick performance of the assay and easy interpretation. One lab from Japan is using nested PCR amplification and sequencing of the Orf1a and S genes as well.
Figure 1. The COVID-19 genetic structure is abbreviated above with the different genes targeted displayed. The names of institutions that have published their assay procedure along with the TaqMan reagents that were reportedly used with each assay are shown above. Primers are represented by small arrows with a TaqMan probe in the middle represented by a black line with green and blue circles indicative of the fluorescent molecule and its quencher. The double set of arrows for the Japanese assay represents a nested PCR strategy.
In silico Cross-reactivity:
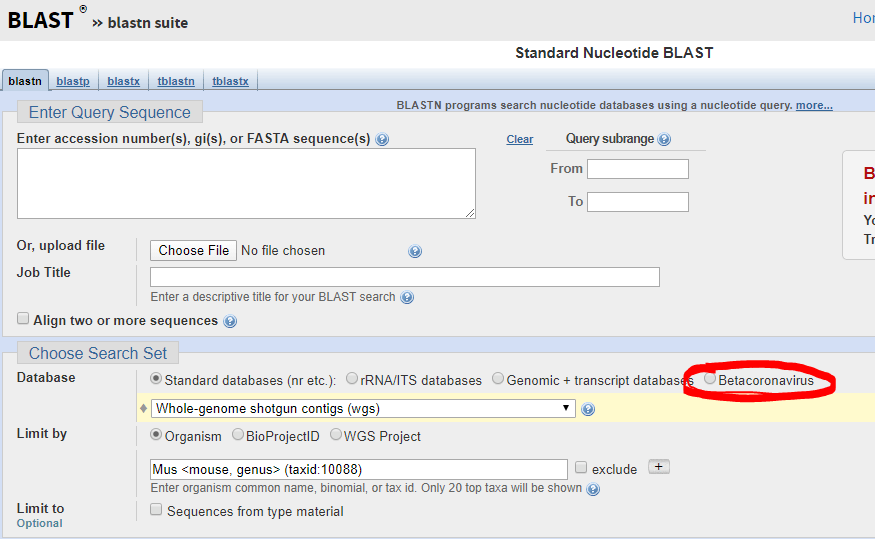
The FDA guidance allows cross-reactivity to be minimally assessed in silico by demonstrating “greater than 80% homology between primer/probes and any sequence present in the targeted microorganism.” The primer locations can be found in the publication of each protocol (except Thermo) and can be confirmed by checking the NCBI Blast site and they actually have a selection for beta-cornavirus (Figure 2) now that allows you to search for your primer’s reactivity across other related viruses- Very helpful!
Figure 2. Select Betacornavirus before entering your primer/probe sequence to confirm cross-reactivity.
Primer/Probe Design:
The N region is the most popular site to probe and is included in most kits once and the CDC kit three times. It was the reagent set for N3 in the CDC kit that was having difficulties, so you may decide to not include that component in your LDT. If you want to see how the different available primer sets align on the N gene sequence you can see below for the primers labeled based on their source. Many are overlapping, perhaps because many people thought the same site was a good target (Figure 3).
Figure 3. N-gene of COVID-19 along with labeled primers from some published assays. The information on the source of the sequence is shown on the bottom right with the link.
Commercially Available Assays:
An important part of validating your COVID-19 assay is to do so quickly. Thus commercially available kits would be helpful, however there are only two commercially available sources at this time: IDT and Thermo. IDT is producing a kit with the CDC design. Thermo produced their kit over the last few months and does not have any published validation information that I could find. Also Thermo when I checked just now for the catalog number, it says this product is unavailable… not sure what that means, but maybe you can try contacting them. Both IDT and Thermo list control plasmid reagents for their assays.
Controls for the Assay:
The wording of the FDA announcement was interesting in that it 1) did not require clinical samples, but allows “contrived clinical specimens.” “Contrived reactive specimens can be created by spiking RNA or inactivated virus into leftover clinical specimens.” A major difficulty is the access to actual COVID-19 RNA or inactivated virus. I noticed that the guidance didn’t say that the assay MUST use RNA. Thus most labs would have access to plasmid DNA, which could potentially be used.
Given the limited availability of RNA for validation use, a lab may consider performing much of the assay optimization with COVID-19 Plasmid DNA while waiting for access to RNA. I would like to be sure my assay could extract, amplify and detect RNA as part of the clinical validation.
Asuragen can produce Armored RNA, with synthetic RNA packaged inside of a viral capsid, which would be a useful control for extraction, amplification and detection. However, we heard this will not be available for another month.
Tom Stenzel (director of the Office of In Vitro Diagnostics and Radiological Health at the FDA’s Center for Devices and Radiological Health (CDRH)) said FDA, BARDA, and the CDC will prioritize and coordinate shipments of viral materials to labs when they are ready to validate tests according to a webinar with labs on Monday. Currently, the FDA is directing inquiries to BEI, which is reportedly prioritizing requests to send out samples in 12-72 hours.
Lastly, one could try to use in vitro synthesized RNA sequences surrounding your primer targets as a control for now and may have better luck in getting the product soon. This is the control that is being shipped with the CDC kits to public labs.
Limit of Detection is an unknown for what is likely to be clinically relevant as we don’t know what the levels look like in people with early vs. late vs. severe vs. mild disease. The FDA just says you should be able to detect 95% of samples (19 of 20) that are x1-x2 the limit of detection.
FDA Notification:
This is the final and important step. Once you go live, you must notify the FDA with an Emergency Use Assay (EUA) form within 15 days. Reviewing the form, there doesn’t appear to have complex explanations or overdue requirements for reporting, which wouldn’t be found in a standard lab validation document.
Final Thoughts/Future commercial solutions:
This information is the best of what I know right now based on current information- this is not a complete guide and the FDA guidance should be read closely for all compliance details.Information is changing quickly and is likely to change more if the number of COVID-19 cases in the United States increases. Cepheid, Luminex, and BioFire are reportedly working on assays that will be out in several months and would be easy to use for many labs that already have one or both of these systems-however it may require a full validation for an LDT, but I’m not sure as it is an EUA-further clarification on this point is needed. Although there are several commercial solutions available, we don’t know how demand could impact supply from each company. Fortunately, some large reference labs like LabCorp and Quest are looking to develop a COVID19 test. Good luck, stay safe, and feel free to contact me with any questions in the comments below so that everyone can benefit from the discussion!
References
In lieu of a list of references, I’ve included web links for the most current and direct sources of information.
-Jeff SoRelle, MD is a Chief Resident of Pathology at the University of Texas Southwestern Medical Center in Dallas, TX. His clinical research interests include understanding how the lab intersects with transgender healthcare and improving genetic variant interpretation.