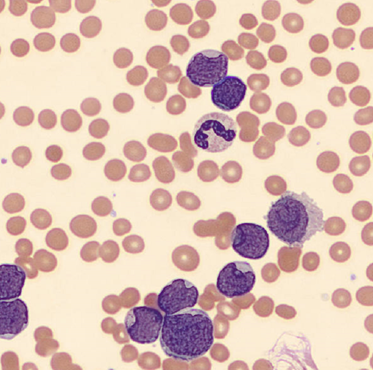
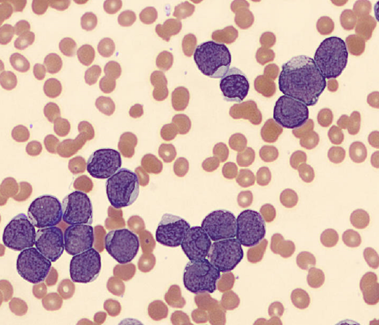
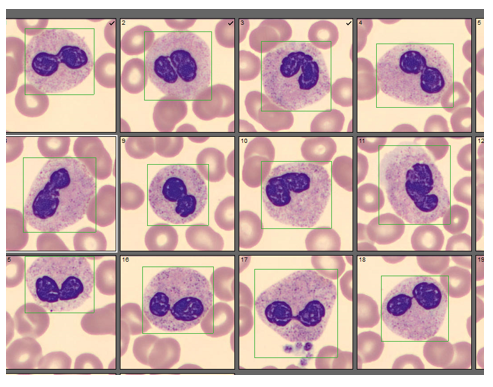
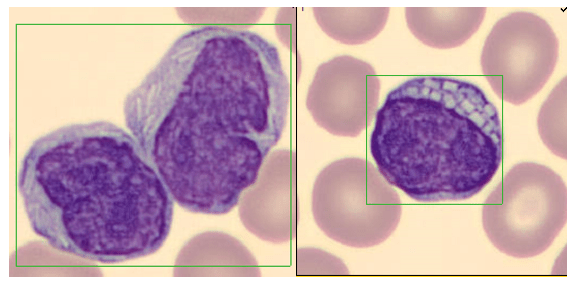
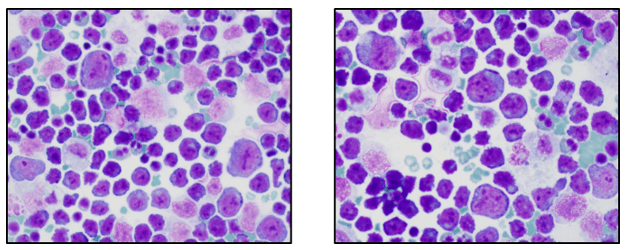
One of the reasons I love working in Hematology is because when we have unexpected results they are often accompanied by visuals… and a picture is worth a thousand words! Unusual or critical results in Chemistry can be interesting, sometimes there are dilutions to perform, results to compare or puzzles to solve, I have always loved working up a good antibody or complicated multiple antibodies in Blood Bank or calculating how many units I may need to screen to find compatible ones, gram stains of unusual organisms in Microbiology can be exciting, but nothing beats some of the cells we see in Hematology! It’s always fascinating when we find unusual cells and follow up with smear reviews with our pathologists. And, being able to save these visuals in CellaVision or saving the slides for teaching, is a plus. These cases are a gift that keeps on giving! Lately I’ve had my share of “exciting” specimens, usually on a Saturday or Sunday afternoon! It never fails to get the adrenaline going when you are the first one to see a CBC with a WBC of 400,000, a differential that is over 90% blasts, rare lymphoma cells, malarial parasites, or a body fluid smear full of malignant cells. The following 2 cases are a very remarkable looking smear and a not so remarkable one, from 2 different patients with the same diagnosis.
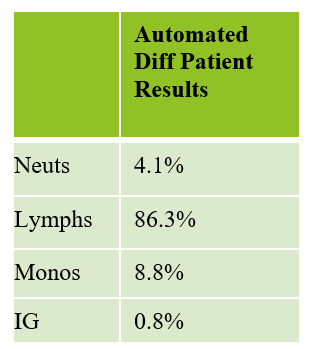
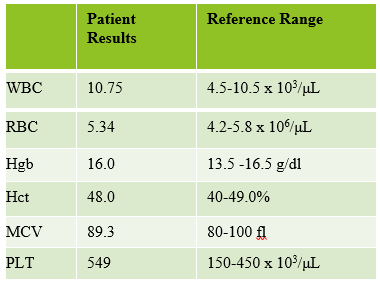
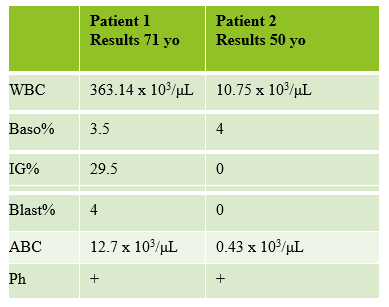
The first patient is a 71 year old male who had a routine CBC done by his primary care physician. The blood was collected as an outpatient on a Saturday morning, and brought to our lab by a routine courier that afternoon (of course, right before change of shift!). We had one previous CBC result on this gentleman, from several years earlier, which was essentially normal. CBC result shown below:



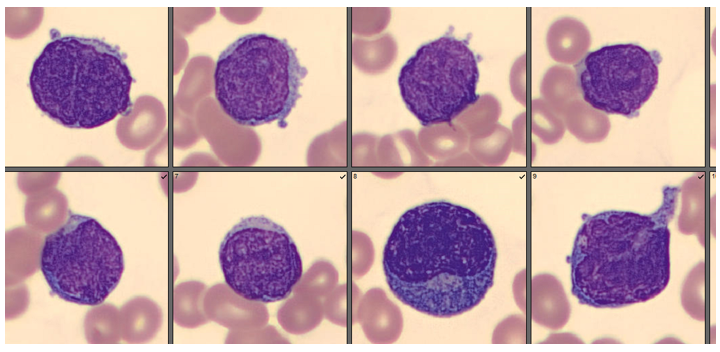
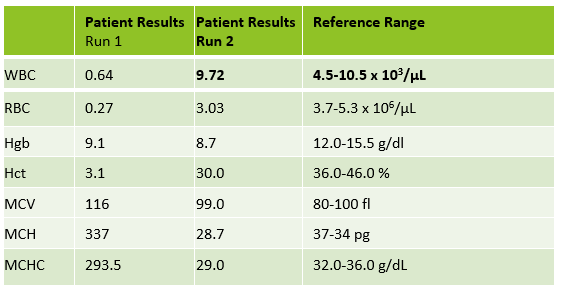
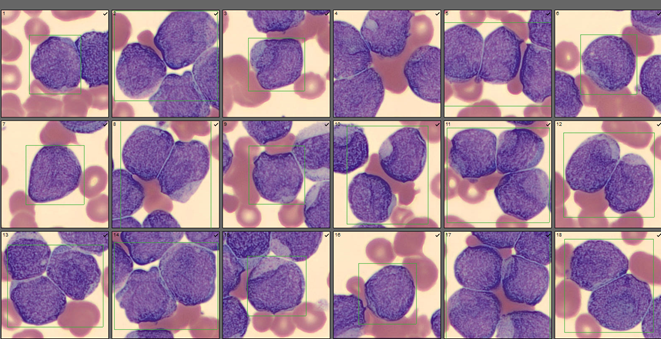
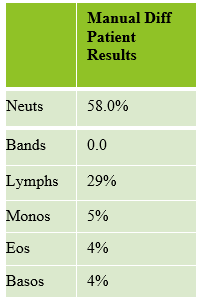
As soon as I saw the results, I called the provider with the WBC and alerted them that I would be contacting the pathologist on call and calling back with the differential. Our pathologist confirmed blasts on the peripheral smear and requested that the sample be sent out for flow cytometry. The pathology report stated “Marked leukocytosis with left shift and <5% blasts. The presentation is suspicious for a myeloproliferative neoplasm (e.g. chronic myelogenous leukemia (CML)). Immunophenotypic studies have been ordered and will be reported separately. Clinical correlation and Hematology consult recommended.” Flow cytometry results showed left shifted maturation and FISH studies demonstrated t(9;22) BCR-ABL with 98% of positive nuclei in bone marrow. No other mutations were detected. Diagnosis: chronic myelogenous leukemia. Five days later, we had a bone marrow scheduled on a 50 year old male. A CBC done 2 weeks earlier showed a mild leukocytosis and thrombocythemia. (WBC 12.4, Hgb 17.8, Hct 52%, PLT 539). Diagnoses under consideration were possible CML, polycythemia or a myeloproliferative neoplasm (MPN). The patient’s CBC the day of the procedure is shown below.
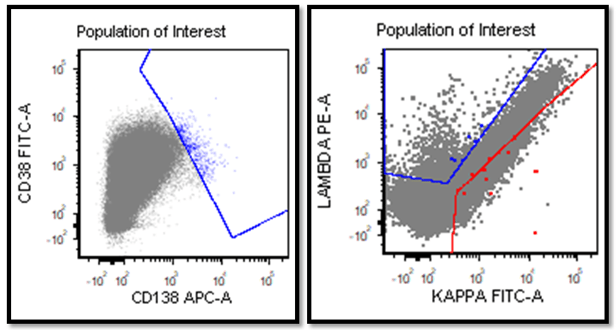
Cytogenetic analysis showed an abnormal clone characterized by the Philadelphia chromosome translocation, t(9;22). The BCR/ABL gene rearrangement was detected by FISH, with 78% of positive nuclei in bone marrow. The bone marrow was negative for other mutations. Flow cytometry analysis reported no evidence of abnormal myeloid maturation or increased blast production. There was no evidence for a lymphoproliferative disorder. Diagnosis: chronic myelogenous leukemia.
In 1959, at a time when techniques for preparing chromosomes for visualizing under the microscope were still very unsophisticated, 2 researchers in Philadelphia detected a tiny abnormality in the chromosomes of patients with CML. They noticed that part of chromosome 22 appeared to be missing. It was not until 1970, when techniques for chromosome banding became available, that this discovery was shown to be a translocation between chromosomes 22 and 9. The shortened chromosome 22 was named the Philadelphia (Ph) chromosome after the city where it was discovered.
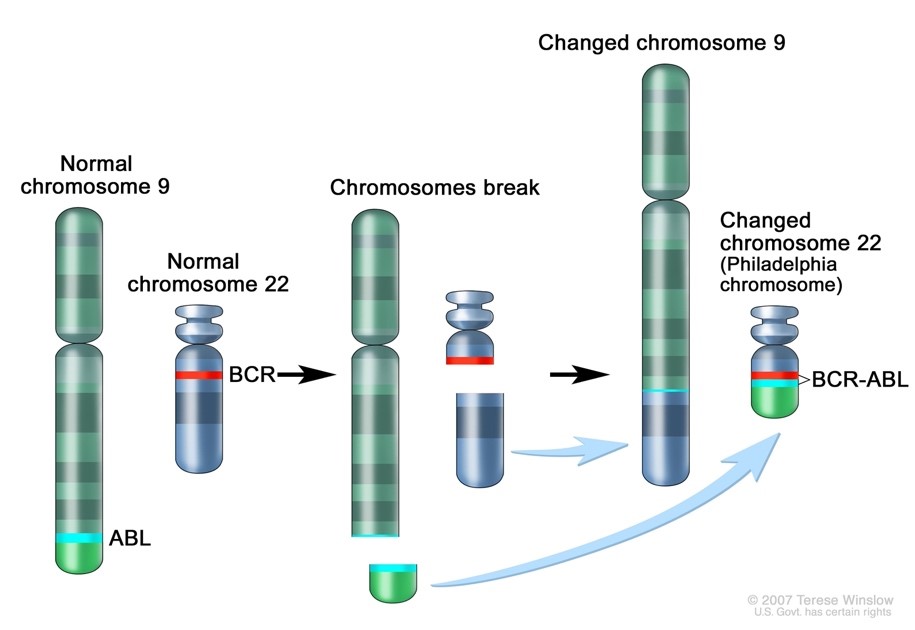
At diagnosis, over 90% of CML cases have the t(9;22) translocation which has become a hallmark for a diagnosis of CML. However, the Ph chromosome is also detected in about 30% of adult acute B cell lymphoblastic leukemia (B-ALL), and a very small number of acute myeloid leukemias (AML) and childhood B-ALL so testing must be done for differentiation. t(9;22) is a translocation of the proto-oncogene tyrosine-protein kinase ABL1 gene on chromosome 9 and the breakpoint cluster region BCR gene on chromosome 22. The newly formed chromosome 22 with the attached piece of chromosome 9 is called the Philadelphia chromosome. The BCR-ABL oncogene is formed on the Philadelphia chromosome and the product of the Ph translocation is an abnormal fusion protein, p210, which has increased tyrosine kinase activity. This, in turn, is responsible for the unregulated proliferation of cells seen in CML. Tyrosine kinase inhibitors (TKIs) have been developed as targeted therapy for Ph+ CML.1
So, how can these 2 patients with very different CBC results both be diagnosed with CML? CML can be classified into phases of CML-chronic phase (CML-CP), accelerated phase (CML-AP), and blast crisis (CML-BP). The WHO Classification of 2017 proposed a system of cutoffs to define each phase. The phases are based mainly on the number of blasts in the blood or bone marrow. Progression from CML-CP to CML-AP is also generally recognized to correlate with an increase in BCR-ABL1 levels. Several studies have been done that discuss another phase, pre-leukemic (pre-clinical) CML. These pre-leukemic patients have the Philadelphia chromosome, the genetic hallmark of CML, without other abnormalities. They have a normal to mildly elevated WBC and are asymptomatic. In these cases, progression to CML-CP can be several months to several years. One interesting factor common in this phase, which can help in diagnosis, is the presence of an absolute basophilia (ABC) >200/mm3. This basophilia is also seen in CML-CP and often progresses with the disease.2
Results from both patients are compared below. While we may more readily recognize a new CML that presents with very high WBC, left shift, and blasts, FISH, flow and cytogenetics of both these patients indicated a diagnosis of CML. This second patient may be one that could be classified as a pre-CML. The patient is certainly fortunate to have physicians who suggested further workup so he can benefit from his early diagnosis.
References
- Huma Amin*, Suhaib Ahmed. Characteristics of BCR–ABL gene variants in patients of chronic myeloid leukemia. Open Medicine, 2021.16:904-912.
- Aye, Le Le; Loghavi, Sanam; Young, Ken H et al. Preleukemic phase of chronic myelogenous leukemia: 2. morphologic and immunohistochemical characterization of 7 cases Annals of Diagnostic Pathology. April 2016 21:53-58 Language: English. DOI: 10.1016/j.anndiagpath.2015.12.004.
- Kuan JW, Su AT, Leong CF, Osato M, Sashida G. Systematic review of pre-clinical chronic myeloid leukaemia. Int J Hematol. 2018 Nov;108(5):465-484. doi: 10.1007/s12185-018-2528-x. Epub 2018 Sep 14. Erratum in: Int J Hematol. 2018 Nov 7;: PMID: 30218276.
- https://www.cancer.gov/publications/dictionaries/cancer-terms/def/philadelphia-chromosome

-Becky Socha, MS, MLS(ASCP)CMBBCM graduated from Merrimack College in N. Andover, Massachusetts with a BS in Medical Technology and completed her MS in Clinical Laboratory Sciences at the University of Massachusetts, Lowell. She has worked as a Medical Technologist for over 40 years and has taught as an adjunct faculty member at Merrimack College, UMass Lowell and Stevenson University for over 20 years. She has worked in all areas of the clinical laboratory, but has a special interest in Hematology and Blood Banking. She currently works at Mercy Medical Center in Baltimore, Md. When she’s not busy being a mad scientist, she can be found outside riding her bicycle.