Too many platelets? We know that low platelet counts can pose problems for hematology analyzers and that reporting accurate results is vital for good patient care. We learn and read a lot about thrombocytopenia and its various symptoms, causes, and treatments. But, what about thrombocytosis? What happens when there are too many platelets?
In my last blog I compared 2 cases of newly diagnosed CML. Lately I have seen so many new leukemia cases and myeloproliferative diseases that I have become fascinated with them. When I was in college and grad school (many moons ago), nomenclature, diagnoses and knowledge of these disorders were very different, so it’s been fun learning about them all over again!
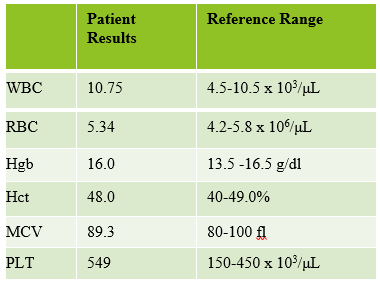
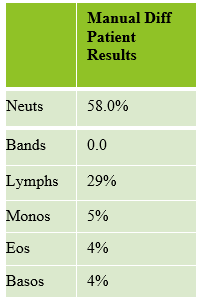
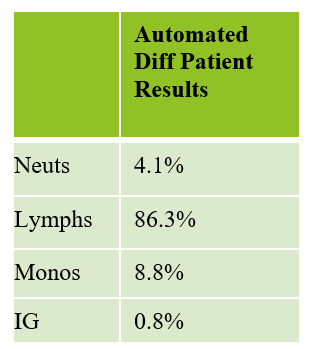
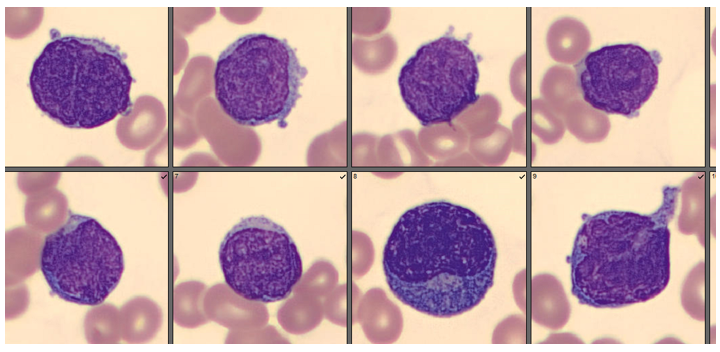
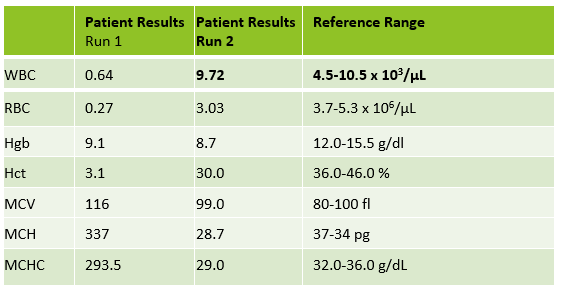
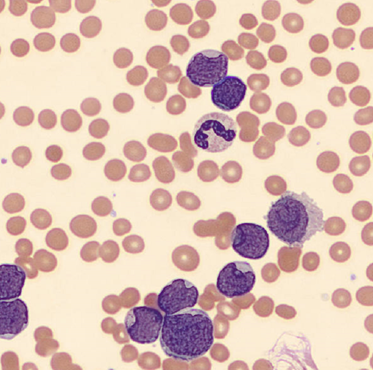
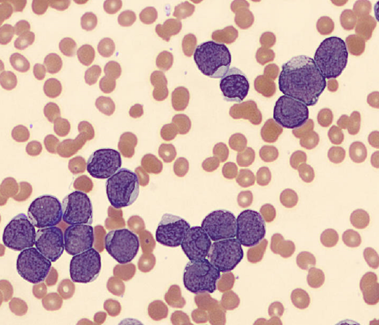
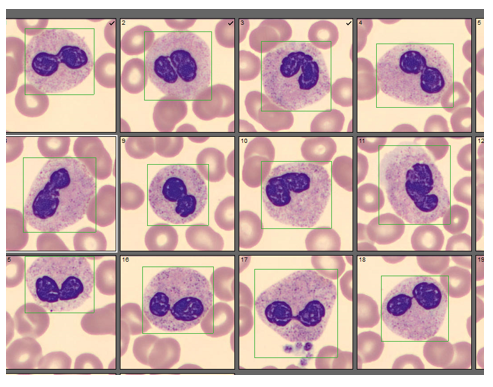
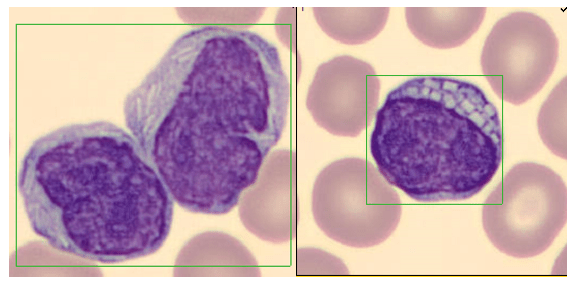
Today’s case is of a 55 year old woman who was referred for a hematology consult because of a finding of increased RBC and platelet counts. White blood cells appeared normal with few reactive lymphocytes noted. The peripheral smear showed mild anisocytosis and dacrocytes. Platelets were markedly increased with large forms present. No giant platelets were noted. A bone marrow biopsy was ordered. Pre-Op diagnosis: Thrombocytosis.
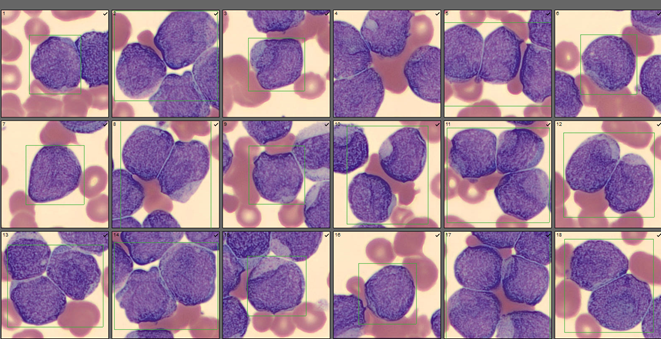
Bone marrow results reported increased myeloid forms with full spectrum of maturation, erythroid elements normal in number with normoblastic maturation, and markedly increased megakaryocytes with numerous large hyperlobated forms. M:E ratio was increased. No iron was seen on iron stain. A reticulin stain showed mildly increased reticulum fibrosis (1+). Next generation sequencing studies demonstrated a JAK2 V617F mutation. BCR-ABL mutation was not detected. Diagnosis: Myeloproliferative neoplasm most consistent with Essential Thrombocythemia (ET).
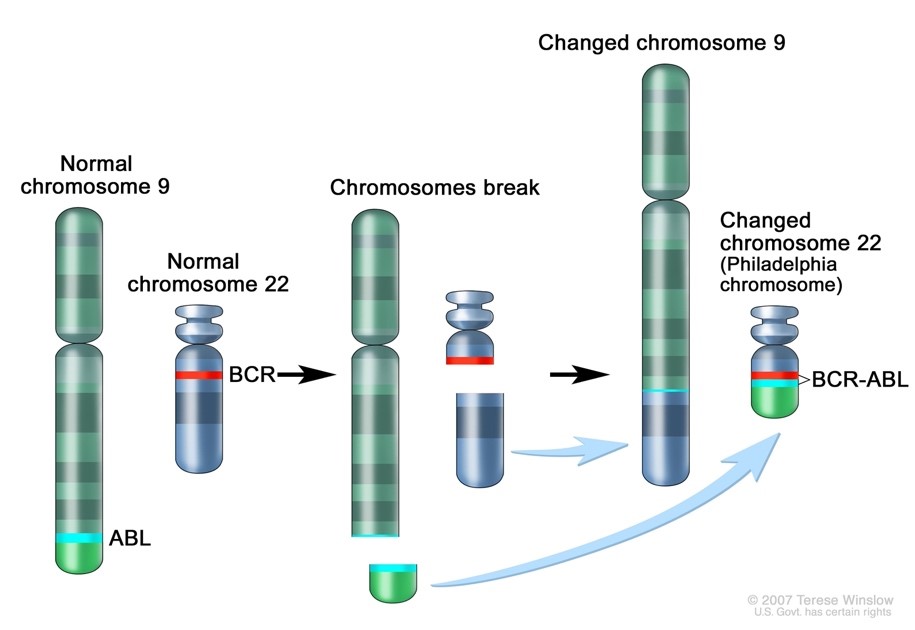
Myeloproliferative neoplasms (MPN) are a group of disorders characterized by the over proliferation of WBCs, RBCs, or platelets. These can be separated into the Philadelphia chromosome (Ph) positive Chronic Myelogenous Leukemia (CML) and Ph negative neoplasms. The BCR-ABL oncogene is formed on the Ph and is responsible for the unregulated proliferation of cells seen in CML. At diagnosis over 90% of CML cases are BCR-ABL positive. (See Case Studies in Hematology: Presenting a double feature starring Chronic Myelogenous Leukemia). On the other hand, Polycythemia Vera (PV), Essential Thrombocythemia (ET) and Primary Myelofibrosis (PMF) are the three classic Ph negative neoplasms.
Many ET patients have no symptoms at diagnosis but are found to have a high platelet count on a routine CBC. Diagnosis is based on ruling out other disease and testing for genetic mutations, which can be done from a peripheral blood sample or bone marrow. In addition to any blood tests, a bone marrow biopsy is typically recommended for differential diagnosis of MPNs. The most common mutation found in PV, ET or PMF, is in the JAK2 gene. The JAK2 V617F mutation is found in nearly all PV patients, and about 50-60% of ET and PMF patients.Othermutations found in the classic MPN group include CALR, the second most common genetic abnormality after JAK2 mutations,and MPL W515L.Normally, blood cells are only produced when the body has a need for the cells, but these genetic mutations turn a gene ‘on’, causing the unregulated production of the affected blood cell line. Until recently it was believed that a patient with PV, ET or PMF will have a mutation in only one of these genes. However, in 2018, a French group reported that CALR or MPL mutations may co-exist in a small percentage of patients with a low burden of JAK2 V617F mutation. (Accurso) Some patients are triple-negative for the JAK2, MPL and CALR mutations and always have a poor prognosis.
The identification of a genetic marker in MPNs is valuable because a JAK2 mutation distinguishes PV from other disorders that may cause polycythemia. As well, a JAK2 or other mutation can distinguish ET from other causes of reactive thrombocytosis and PMF from secondary causes of myelofibrosis. In addition, most CML cases are diagnosed with a very high WBC, but occasionally patients with CML have a normal or only slightly elevated WBC with a high platelet count. Therefore, patients with suspected ET are also evaluated for CML with a test for the Philadelphia chromosome. Our patient was found to have a JAK2 V617F mutation, BCR-ABL negative and was diagnosed with ET.
ET was first recognized in the 1950’s and was termed a myeloproliferative disorder. At this time, it was not known what was causing the over proliferation of platelets. Theories were broad and ranged from ‘something environmental’ to ‘an internal defect’. Over the decades, it became more apparent that the myeloproliferative disorders were caused by internal defects in stem cells, and they were renamed MPN. In 2005, four separate research groups, using different methods, all identified the JAK2 V617F allele, which led to further understanding of PV, ET and MPN. The MPL mutation was discovered in 2006 and CALR mutations were discovered in 2013.
ET is a type of chronic leukemia and patients with ET generally have a normal life expectancy. Of the 3 BCR-ABL negative MPN, ET has the best prognosis. Treatment is often not needed, other than aspirin for prevention of blood clots. Patients are placed in risk factor groups based on risk of clots or bleeding. A patient <60 years with no JAK 2 mutation and no prior thrombosis is considered very low risk and would be simply observed or prescribed low dose aspirin. Patients <60, JAK2 V617F +, with no prior thrombosis have low risk and would be treated with aspirin, dosage dependent on any cardiac risk factors. Older patients over 60 with JAK2 wild type and no history of thrombosis may be treated with aspirin alone or with cytoreductive therapy. Lastly, the highest risk patients are those over 60, JAK2 V617F + or with prior thrombosis, and would be treated with cytoreductive therapy, such as hydroxyurea. With very high platelet counts, there is a risk of both blood clots and hemorrhage. Blood clots that develop in thrombocythemia can use up the body’s platelets and result in bleeding. For this reason, cytoreductive therapy such as hydroxyurea is recommended to reduce hemorrhage in high-risk patients with very high platelet counts over 1,000 x 103/ μL. Hydroxyurea can also be used as treatment in patients who have a mixed population of PV and ET. CALR mutated patients with ET tend to be young with a much lower thrombotic risk and do not generally require therapy. Aspirin in this group is considered overtreatment because CALR+ patients suffer more risk of bleeding with aspirin.
While there is some risk of a MPN transforming to another type, ET is the MPN least likely to transform or to progress to acute myeloid leukemia. ET also has a better prognosis than the other MPN. Even so, there is often not one clear cut entity. There can be overlap between the disorders, causing some difficulty in diagnosis and treatment decisions. For instance, a physician may have a patient, as our patient does, with a high RBC and Hgb, with thrombocytosis, and with a JAK2 mutation. Bone marrow biopsy may detect hyperlobated megakaryocytes which would indicate a diagnosis of ET; however, the physician may choose to monitor and possibly treat as PV due to the RBC counts and symptoms.
Many advances in the understanding of ET and molecular techniques for diagnosis have been made in the last 10 years. Unfortunately, many times, diagnosis is not made until after a thrombotic event. In addition, many patients with thrombocytosis are not referred for hematology consults in a timely fashion or until they too experience a thrombotic event. In 2016 WHO published a new diagnostic criterion for PV, ET and PMF. There is an effort amongst research and physician groups to ‘spread the news’ throughout the medical community to promote early detection of ET, minimize the risk of thrombotic events and improve prognosis.
References
- Accurso V, Santoro M, Mancuso S, et al. The Essential Thrombocythemia in 2020: What We Know and Where We Still Have to Dig Deep. Clin Med Insights Blood Disord. 2020;13:2634853520978210. Published 2020 Dec 28. doi:10.1177/2634853520978210
- Bose P, Verstovsek S. Updates in the management of polycythemia vera and essential thrombocythemia. Ther Adv Hematol. 2019;10:2040620719870052. Published 2019 Aug 30. doi:10.1177/2040620719870052
- Kilpivaara, O., Levine, R. JAK2 and MPL mutations in myeloproliferative neoplasms: discovery and science. Leukemia 22, 1813–1817 (2008). https://doi.org/10.1038/leu.2008.229
- Panjwani,Laura. Management of ET, PV Requires 2 Distinct Approaches. Special Reports, Hematologic Malignancies: Polycythemia Vera, Volume 3, Issue 3. September 28, 2016
- https://rarediseases.org/rare-diseases/essential-thrombocythemia/
- https://www.mpnconnect.com/pdf/who-diagnostic-criteria-mf-pv-et.pdf

-Becky Socha, MS, MLS(ASCP)CMBBCM graduated from Merrimack College in N. Andover, Massachusetts with a BS in Medical Technology and completed her MS in Clinical Laboratory Sciences at the University of Massachusetts, Lowell. She has worked as a Medical Technologist for over 40 years and has taught as an adjunct faculty member at Merrimack College, UMass Lowell and Stevenson University for over 20 years. She has worked in all areas of the clinical laboratory, but has a special interest in Hematology and Blood Banking. She currently works at Mercy Medical Center in Baltimore, Md. When she’s not busy being a mad scientist, she can be found outside riding her bicycle.