Why is storytelling so important? People have been doing it for thousands of years so there must be something behind this practice. Storytelling can be a form of entertainment, such as in plays, operas, and even movies. Passing down stories has been responsible for keeping many cultures alive. Think of a group of early homo sapiens gathered around a campfire, telling the tale of a caveman who ate a smooth, round, red berry and they did not wake up the next day. Stories have also been a part of our advancement as humans, and as a society. Without storytelling, early civilizations may not have been able to farm, build cities, or even navigate the open oceans. When it comes to laboratory safety, stories are also very valuable and should be shared for several reasons. These stories help employees and staff stay safe, but equally as important, they can protect patients. Safety stories not only shed light on the dangers found in the laboratory but can also strengthen the lab’s safety culture by igniting conversations around safety withing the team itself.
So, what kinds of laboratory safety stories can be shared? The most obvious might be stories involving employee injuries or exposures. This type of safety story really has two purposes. First, it can generate discussion around how the injury could have been avoided. If a coworker sustained a hazardous chemical splash to the face, but was wearing safety goggles, staff should conclude that full-face shields must be worn for that process to provide adequate protection. Secondly, the story can make the possibility of an incident more of a reality to others not involved. Have you ever heard someone say, “that could never happen in my lab?” Telling true stories about incidents raises awareness that those types of things can and do happen.
Probability and science demonstrate that almost every possible accident can happen in a lab, it just has yet to occur. Without events occurring over time, employees may become complacent with the biological and chemical risks in the lab. Once this feeling of “immunity to incidents” sets in, employees begin to take short cuts or to pay less attention to tasks, then the chances of an accident increase. Something else could make someone deliberately let their guard down. A person may choose to perform an unsafe act because they do not perceive a risk associated with their actions. If an individual feels that the risk is low or absent, they may not take the necessary precautions (like donning their PPE) before answering a lab phone or pouring xylene into a beaker. Sharing the details around lab accidents will help eliminate any false feelings of security staff may have when working with hazardous substances.
Safety stories do not always have to be centered around negative outcomes, though. Safety stories can also highlight near misses or great catches. It is just as important to discuss (or even celebrate) when bad outcomes are avoided. Talking about events with a positive spin can help motivate staff and boost morale. Some safety stories may not be directly lab-related. It might be just as meaningful to share a story about walking out of the office without your keys and getting locked out of the department.
When is it a good time to share a safety story? Luckily, I work for an organization that believes in starting every meeting with a safety story. Each morning, managers from all hospital locations, along with other key support staff, meet virtually to discuss important items and the status of their labs. Before the team members report out, the person leading the meeting will ask a participant to share a safety story. Of course, injuries and exposures are shared, but other times team members may talk about a successful conversation they had with an outside department. Recently, when an unusual amount of snow fell in the state, many safety stories were shared about how to safely walk on ice and why it might not be a good idea to use hot water to help defrost your car windshield.
It is great that the managers get a chance to share these stories, but it should not stop there. The safety stories have a greater impact in the lab if the lessons learned make it to the staff level. Sharing these stories at a shift change or huddle can be very helpful. Labs with the greatest safety culture are those that routinely discuss safety daily.
One goal of sharing safety stories in the lab is to ultimately avoid unfavorable events. To accomplish this goal, the lab must first understand how the event occurred and what might be missing from the current process that created the opportunity for failure. Like the stories of old that helped keep cultures alive for generations, safety stories are an awesome tool that will maintain your own strong safety culture for years in the laboratory.
-Jason P. Nagy, PhD, MLS(ASCP)CM is a Lab Safety Coordinator for Sentara Healthcare, a hospital system with laboratories throughout Virginia and North Carolina. He is an experienced Technical Specialist with a background in biotechnology, molecular biology, clinical labs, and most recently, a focus in laboratory safety.
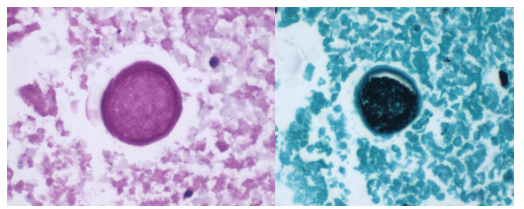
A 58-year-old woman with past medical history significant for degenerative joint disease and an approximately 30-pack per year smoking history was referred for low-dose computed tomography (CT) scan by her primary care provider to screen for lung cancer. An 8-mm nodule was observed in the lingula of the left lung. Follow up PET scans showed mild hypermetabolic activity in this nodule, as well as in axillary and subpectoral lymph nodes. The patient underwent a left video-assisted thoracic surgery to remove the nodule, and intraoperative frozen sectioning of the nodule showed necrosis but was negative for malignancy. On further discussion with the patient, it was discovered that she had spent several years living in Arizona and had participated in outdoor activities but had never observed any acute or chronic respiratory symptoms. On hematoxylin and eosin (H&E) staining of the nodule from biopsy, necrotizing granulomas and endospore-containing spherules were noted. These structures, consistent with Coccidioides, were also visualized under Gomori methenamine silver (GMS) and periodic acid-Schiff (PAS) staining, Of note, tissue culture of the specimen was negative after eight weeks (image 1).
Image 1. Lung tissue from the patient revealed spherules stained with hematoxylin and eosin (H&E) (left) and Gomori methenamine silver (GMS) (right).
Discussion
Coccidioides species, particularly C. immitis and C. posadasii, are dimorphic fungi endemic to the Western hemisphere and have been documented in North, Central, and South America. Within the United States, Coccidioides has historically been associated with the arid southwestern states, with the Centers for Disease Control and Prevention (CDC) noting that many cases of coccidioidomycosis originate in California and Arizona.1 However, both the CDC and a 2022 study evaluating rates of coccidioidomycosis diagnosis have identified cases across the country, most frequently among travelers to high-prevalence areas. In the United States, other regions of endemicity include southwestern New Mexico, Utah, Washington, and western Texas.1,2 This fungus grows best in soil as mycelia, which, in dry environments, develop into spores.3,4 These spores can be released into the air with disruption of the soil, through natural or man-made forces causing infection via inhalation. Generation of dust clouds such as through natural occurrences of earthquakes and windstorms have led to outbreaks and lead to increased risk of exposure for individuals around the area.1,2
Coccidioidomycosis, also called “valley fever”, is a syndrome characterized by pulmonary and systemic symptoms, though it is more frequently asymptomatic. The incubation period is typically 1 to 3 weeks. Patients frequently present several weeks after exposure with fever, chills, night sweats, cough, and arthralgias.4 Given the general nature of these symptoms, it is believed that many cases are undiagnosed or misattributed to other causes of community acquired pneumonia. Mild pulmonary coccidioidomycosis is frequently self-resolving over the course of weeks to months, but persisting respiratory infection in patients previously thought to have bacterial or viral pneumonia can be the first indication of a fungal etiology.3 A small percentage of affected patients can develop more complex disease, including more severe pulmonary involvement and disseminated infection. Complex pulmonary coccidioidomycosis is characterized by pleural effusion, nodules, and cavities. Empyema can occur as a consequence of fistula formation between a peripheral cavitary lesion and the pleural space.3 Disseminated disease has numerous manifestations, including cutaneous and soft tissue disease, osteomyelitis, synovitis, and meningitis.
Coccidioides can also be isolated from respiratory specimens, tissue biopsies and cerebrospinal fluid using routine fungal culture medium. Direct staining for the fungal elements (and sometimes spherules) from unfixed specimens can be done using calcofluor white, fluorescent stain. Procedures that involve manipulation of the sporulating dimorphic molds such as Coccidioides require biosafety level 2 practices and facilities and should be performed in a class II biological safety cabinet. Lifting of the culture plate lid is sufficient to release numerous spores into the environment capable of causing an infection.4 Cultured Coccidioides poses an occupational risk, as there have been reports of laboratory personnel developing symptoms of infection after working with the cultured fungus. As with other dimorphic molds, these organisms exhibit two forms: the mold form grown on Sabouraud dextrose agar or potato dextrose agar grown at 25C and a yeast form when grown at 37C. Mycelia are often visible within the first several days of incubation at 25C. The mold colonies may be white to grey, and sometimes tan or red with age. Microscopic examinations may reveal septate, hyaline, hyphae. Coccidioides form arthroconidia which are one-cell length, cylindrical to barrel-shaped with thick, smooth walls in their hyphae. The arthroconidia may alternate with empty disjunctor cells, giving the impression of alternating staining hyphae. True arthroconidia like Coccidioides will fragment. At 37C, Coccidioides develop into spherules, thick-walled structures where numerous endospores develop. The spherule form is rarely isolated in cultures but can be visualized on PAS, GMS, or H&E stains from fixed tissues.4
Diagnosis can also be made using serological testing, including enzyme immunoassay, immunodiffusion, and complement fixation IgM and IgG assays. These tests have varying sensitivities and specificities, often depending on the manufacturer of the test, though complement fixation and immunodiffusion methods in general have high sensitivities, rendering a positive result as virtually diagnostic.3 Coccidioidin is typically the antigen from the mycelial phase of the mold that is used for serological tests. A positive result from an immunodiffusion complement fixation test typically is indicative of a recent or chronic infection with IgG antibodies detected 2-6 weeks after onset of symptoms. On the other hand, the complement fixation test becomes positive 4-12 weeks after infection. Evaluating the titers can help determine the time and severity of infection. A titer <1:4 suggest early to residual disease whereas titers >1:16 could suggest disseminated disease beyond the respiratory tract.6 Any detection of antibodies from CSF is suggestive of meningitis caused by Coccidioides. Of note, in scenarios where there are discrepant results between serological tests, the patient should be investigated for histoplasmosis or blastomycosis. Antigen detection for Coccidioides species have been developed but performance is poor in urine but slightly higher in CSF specimens; similarly, Beta-D-glucan testing in serum for coccidioidomycosis also has limited value.7,8.9 Molecular methods of diagnosis have also been developed, but only one PCR test has been FDA approved with comparable sensitivity to cultures.10
Treatment of primary pulmonary and disseminated coccidioidomycosis typically consist of the triazoles such as voriconazole, posaconazole, itraconazole, and fluconazole. Amphotericin B can also be used, though this is usually reserved for poor responding or rapidly progressing infection, given its side effects.11 Immunocompromised individuals are at increased risk for disseminated infection whereas immunocompetent individuals may recover without treatment. The Infectious Diseases Society of America notes that mild cases of limited pulmonary involvement can be managed with observation and supportive measures alone.12
2. Mazi PB, Sahrmann JM, Olsen MA, et al. The Geographic Distribution of Dimorphic Mycoses in the United States for the Modern Era. Clin Infect Dis. 2023;76(7):1295-1301. doi:10.1093/cid/ciac882
3. Johnson RH, Sharma R, Kuran R, Fong I, Heidari A. Coccidioidomycosis: a review [published correction appears in J Investig Med. 2021 Dec;69(8):1486. doi: 10.1136/jim-2020-001655corr1]. J Investig Med. 2021;69(2):316-323. doi:10.1136/jim-2020-001655
4. McHardy IH, Barker B, Thompson GR 3rd. Review of Clinical and Laboratory Diagnostics for Coccidioidomycosis. J Clin Microbiol. 2023;61(5):e0158122. doi:10.1128/jcm.01581-22
5. David A. Stevens, Karl V. Clemons, Hillel B. Levine, Demosthenes Pappagianis, Ellen Jo Baron, John R. Hamilton, Stanley C. Deresinski, Nancy Johnson, Expert Opinion: What To Do When There Is Coccidioides Exposure in a Laboratory, Clinical Infectious Diseases, Volume 49, Issue 6, 15 September 2009, Pages 919–923, https://doi.org/10.1086/605441
6. Smith CE, Saito MT, Simons SA. 1956. Pattern of 39,500 serologic tests in coccidioidomycosis. JAMA 160:546–552. doi: 10.1001/jama.1956.02960420026008.
7. Durkin M, Connolly P, Kuberski T, Myers R, Kubak BM, Bruckner D, Pegues D, Wheat LJ. Diagnosis of coccidioidomycosis with use of the Coccidioides antigen enzyme immunoassay. Clin Infect Dis. 2008 Oct 15;47(8):e69-73. doi: 10.1086/592073. PMID: 18781884.
8. Kassis C, Zaidi S, Kuberski T, Moran A, Gonzalez O, Hussain S, Hartmann-Manrique C, Al-Jashaami L, Chebbo A, Myers RA, Wheat LJ. Role of Coccidioides Antigen Testing in the Cerebrospinal Fluid for the Diagnosis of Coccidioidal Meningitis. Clin Infect Dis. 2015 Nov 15;61(10):1521-6. doi: 10.1093/cid/civ585. Epub 2015 Jul 24. PMID: 26209683.
9. Thompson GR 3rd, Bays DJ, Johnson SM, Cohen SH, Pappagianis D, Finkelman MA. Serum (1->3)-β-D-glucan measurement in coccidioidomycosis. J Clin Microbiol. 2012 Sep;50(9):3060-2. doi: 10.1128/JCM.00631-12. Epub 2012 Jun 12. PMID: 22692738; PMCID: PMC3421794.
10. Vucicevic D, Blair JE, Binnicker MJ, McCullough AE, Kusne S, Vikram HR, Parish JM, Wengenack NL. The utility of Coccidioides polymerase chain reaction testing in the clinical setting. Mycopathologia. 2010 Nov;170(5):345-51. doi: 10.1007/s11046-010-9327-0. Epub 2010 Jun 10. PMID: 20535639.
11. Ampel NM. The Treatment Of Coccidioidomycosis.Rev Inst Med Trop Sao Paulo. 2015 Sep;57 Suppl 19(Suppl 19):51-6. doi: 10.1590/S0036-46652015000700010. PMID: 26465370; PMCID: PMC4711193.
12. Galgiani JN, Ampel NM, Blair JE, et al. 2016 Infectious Diseases Society of America (IDSA) Clinical Practice Guideline for the Treatment of Coccidioidomycosis. Clin Infect Dis. 2016;63(6):e112-46. doi:10.1093/cid/ciw360
–Anna Reed is currently a third year medical student at the George Washington University School of Medicine and Health Sciences. She intends to pursue a pathology residency, followed by a fellowship in forensic pathology. Her academic interests include the role of pathology in disaster scenarios, various topics in microbiology, and ways to improve autopsy practices.
-Rebecca Yee, PhD, D(ABMM), M(ASCP)CM is the Chief of Microbiology, Director of Clinical Microbiology and Molecular Microbiology Laboratory at the George Washington University Hospital. Her interests include bacteriology, antimicrobial resistance, and development of infectious disease diagnostics.
The latest cinematic reboot of the Fantastic Four will coming to theaters in 2025. For those paying close attention, this will mark the fourth film featuring the team that effectively started the “Marvel Superhero Age” back in 1961 (Actually, it is the fifth film if you count the 1994 low-budget, never-released version which was only made so the movie company could keep the property rights). None of the previous Fantastic Four movies made a big splash at the box office, which is a shame for a popular comic book that has been in print for over 60 years. Nonetheless, Hollywood is going to give this franchise a recharge, another shot at success. If you’ve ever given up on trying yet again to change your lab’s safety culture, there may be a lesson or two here.
A laboratory’s safety culture isn’t something you can set and forget. Like an instrument that requires calibration or a reagent that needs restocking, the safety culture needs regular attention, especially when changes occur. New managers step in, staff members rotate in and out, and fresh challenges emerge. Over time, what once felt like a strong safety mindset can fade, leaving bad habits unchecked.
Recharging a lagging lab safety culture starts with intentional action—setting new standards, educating staff, and establishing a timeline for change. Whether you’re stepping into a leadership role for the first time or trying to reinvigorate a culture that has grown complacent, there is a way to hit the reset button and energize your lab’s commitment to safety.
When a new manager takes the reins, it’s a prime opportunity to reinforce safety priorities. If you’re that manager, don’t assume that existing safety practices are being followed correctly—take the time to observe, ask questions, and get a sense of what’s really happening in the lab. One of the best ways to establish authority in safety is to lead by example. If staff see their manager wearing PPE properly and promptly addressing safety issues, they’re more likely to do the same. Communicate safety expectations clearly from day one, letting staff know that maintaining a safe work environment isn’t an option—it’s a requirement.
A safety-focused leader also builds relationships. Encourage open dialogue about safety concerns and emphasize that you are there to support—not punish—those who speak up. A strong safety culture depends on trust, and that starts with leadership.
Sometimes, a lab’s safety culture erodes because expectations have become unclear. Maybe PPE compliance has gotten lax, or near-miss reporting has dwindled. If you’re looking to recharge your lab’s safety mindset, now is the time to set (or reset) clear, non-negotiable standards.
Start by reviewing current policies and identifying gaps. Are safety inspections being conducted regularly? Are employees following established chemical hygiene protocols? Once you’ve pinpointed weaknesses, work with your team to set measurable goals for improvement. Accountability is key. Safety should never be an afterthought or a box to check—it should be embedded in daily operations. Reinforce expectations with frequent check-ins, and if noncompliance issues arise, address them immediately with coaching and retraining. Employees need to know that safety rules aren’t just suggestions—they’re essential.
Recharging a lab’s safety culture doesn’t happen overnight. It requires planning, execution, and follow-through. The best way to ensure momentum is to establish a start date for new safety practices and make sure everyone knows it.
Maybe you’re rolling out a new PPE policy, implementing a new safety reporting system, or introducing a fresh set of lab safety goals. Whatever the change, give employees a clear timeline for when new expectations take effect. Set reminders, provide refresher training, and follow up to make sure changes are being implemented. The start date isn’t just about accountability—it’s about setting a tone. It tells staff that safety is a priority, not just an idea.
A strong lab safety culture isn’t a one-time achievement—it’s an ongoing effort. Whether you’re a new leader setting the tone, a seasoned manager reinforcing expectations, or a team member committed to a safer work environment, your actions make a difference.
By addressing leadership transitions, engaging rotating staff, setting clear standards, improving training, and establishing firm start dates, you can recharge your lab’s safety culture and create an environment where safety is second nature.
So take a look around—does your lab’s safety culture need a boost? Have you tried a boost or two in the past that didn’t work? Don’t get tired of trying, even if it’s your fourth or fifth reboot. A renewed look at safety isn’t just about compliance; it’s about ensuring that every technologist, phlebotomist, and laboratory professional goes home safe at the end of the day. A lab that succeeds in this are can only be called…fantastic!
–Dan Scungio, MT(ASCP), SLS, CQA (ASQ) has over 25 years experience as a certified medical technologist. Today he is the Laboratory Safety Officer for Sentara Healthcare, a system of seven hospitals and over 20 laboratories and draw sites in the Tidewater area of Virginia. He is also known as Dan the Lab Safety Man, a lab safety consultant, educator, and trainer.
Despite being a forensic pathologist, I (and many of my colleagues) still consider our work “patient care.” The people we autopsy are our patients, even though they have not actively sought care. Knowing this, forensic pathologists understand that we still must maintain our patient’s dignity and provide compassionate care, even after their death. Not infrequently, they are people who, in life, avoided doctors and hospitals for many reasons – a distrust of mainstream medical establishments, financial insecurity, previous negative experiences with medical care, or a combination of all three. Yet because they have no documented medical history or physician to sign their death certificate, we are the last doctor they see.
Transgender people, unfortunately, often fall into this category. Studies have shown that transgender people are more likely to experience poverty and food insecurity, and they have often had negative experiences while seeking healthcare. There is data to suggest that transgender and other gender diverse individuals are at increased risk of violent death, including homicide and suicide.
Before continuing, it may be helpful to review some definitions. “Sex” refers to the phenotypic markers of one’s chromosomes. In contrast, “gender” refers to a self-assigned identity, based on societal constructs of expected appearances and behaviors. At birth, one’s assigned gender often defaults to the phenotypic sex. As one becomes older, people may begin to realize that their gender identity and manner of expression differs from the gender assigned at birth – and hence, they may self-identify as “transgender” or “non-binary”.
Unfortunately, the available evidence regarding health risks for transgender people is limited by the lack of data collection in death investigation. Death certificates in most states do not offer a “transgender” or “non-binary” option. Legally the physician signing the certificate may be compelled to put the assigned gender at birth, if it was never formally changed on legal identification documents. Some national databases of violent or drug-related deaths include a checkbox to mark an individual as non-binary or transgender, but this may be omitted if the person entering the data is unsure of the definition or is unaware of the decedent’ gender identity. Additionally, transgender people are often misgendered in investigative and media reports. This has been described as a “nonconsensual detransitioning”, which leads some transgender individuals to worry about how they will be remembered after death.
This misgendering and erasure of an individual’s identity contributes to two distinct problems. The first problem is direct harm to the deceased, by disrespecting their self-identity, and contributing to distrust of the medical profession by the transgender community. The second is a lack of quality public health data to enumerate risks and identify where preventative efforts are best focused.
It is only within the past 5 to 10 years that changes are being made to infrastructure systems to better collect this data. There are also increasing efforts to educate death investigators on documenting and asking questions about sexual orientation and gender identity (aka “SOGI”) data. In death, we have an obligation not only to maintain the dignity of our patient, but to collect accurate and complete data to identify existing threats to public health. Government executive orders notwithstanding, transgender and non-binary people exist. And they deserve compassionate and affirming medical care, in both life and death.
REFERENCES
Fedor, Juniper MS, PA (ASCP)CM; Krywanczyk, Alison MD; Redgrave, Anthony EdD. Gender Identity in Forensic Death Investigation: A Narrative Review and Suggested Guidelines for Documenting and Reporting. The American Journal of Forensic Medicine and Pathology 45(3):p 231-241, September 2024.
Blosnich, John R. PhD, MPH; Butcher, Barbara A. MPH; Mortali, Maggie G. MPH; Lane, Andrew D. MSEd; Haas, Ann P. PhD. Training Death Investigators to Identify Decedents’ Sexual Orientation and Gender Identity: A Feasibility Study. The American Journal of Forensic Medicine and Pathology 43(1):p 40-45, March 2022.
Walters, Jaime K. MPH; Mew, Molly C. MPH; Repp, Kimberly K. PhD, MPH. Transgender and Nonbinary Deaths Investigated by the State Medical Examiner in the Portland, Oregon, Metro Area and Their Concordance With Vital Records, 2011-2021. Journal of Public Health Management and Practice 29(1):p 64-70, January/February 2023.
Alpert AB, Mehringer JE, Orta SJ, Hernandez T, Redwood EF, Rivers L, Manzano C, Ruddick R, Adams S, Sevelius J, Belanger E, Operario D, Griggs JJ. Transgender People’s Experiences Sharing Information With Clinicians: A Focus Group-Based Qualitative Study. Ann Fam Med. 2023 Sep-Oct;21(5):408-415. doi: 10.1370/afm.3010.
-Alison Krywanczyk, MD, FASCP, is currently a Deputy Medical Examiner at the Cuyahoga County Medical Examiner’s Office.
When you think of the “glass ceiling,” you typically think of your career, right? I know I thought purely of professional development when I heard that term thrown around. I’m a huge proponent of breaking said glass ceiling in yourself, your team, your organization, your specialty, and the entire field of laboratory medicine. There’s continuing education through our professional societies and ideally through our institutions, and there are advanced degrees, certificates, and on-the-job training. So many tools and resources we can add to our professional toolbox to exceed all expectations, including those we hold over ourselves. But what about the personal glass ceiling? In order to excel in our careers, we have to first excel in ourselves. Let that sink in. I hear other lab professionals say, “I want to be a leader, too, someday.” I promise you, especially as I’ve learned over the past 3 years in an official leadership role, a leader is not shaped without ongoing personal development and the constant challenging of your personal glass ceiling.
New people-leaders in my health system are required to undergo extensive training through our Leadership and Organizational Development (LOD) team. To be quite frank, I silently scoffed at the idea of having to attend “so many virtual and in-person trainings that interfere with my workday. I mean, my dissertation focused on laboratory leadership and change management, come on!” I’ll be the first to admit that I was so wrong. It also helped that there were three other laboratory leaders present with me, so we were able to role play and come up with solutions to issues in real-time. There were DEI trainings, such as civil treatment and inclusive workforce; communication models, including coaching, delegation, and two of my personal favorites – Management Foundations and Emotional Intelligence (EQ).
In the first day of Management Foundations (and yes, after my thinking, “How can I get out of this and prepare for our upcoming lab inspection instead?”), we were given tools to explore ourselves. Initially, I thought we would be completing the Myers-Briggs Type Indicator and explore our personalities. I’m in my seat waiting to share how I’m an INFJ, the Advocate, and how I have (sometimes) unrealistic high expectations for myself and others. This wasn’t a personality test though. What we were about to embark on was a behavior test, also known as the DiSC Assessment. We were told to choose 4 color-coded cards with adjectives that we thought described ourselves. I chose “high standards,” “diplomatic,” “analytical,” and “enthusiastic.” I chuckled when I saw a fellow lab supervisor also grab “high standards” to describe herself as I know how Type A we both are. After going through the formal assessment, I learned that my dominant behavior was “Conscientiousness” with a secondary behavior of “Influence.” As a “C” behavior type, I’m described as enjoying working with people who are organized and have high standards, carefully weighing pros and cons, and preferring environments with clearly defined expectations. Nailed it! This assessment teaches you how to communicate and work with different behaviors and how those other behaviors react to pressure. It is also important to take note of your secondary behavior as sometimes that can become the dominant behavior under stress. This activity was so enlightening that I returned to the lab the following day and asked my team complete the assessment as well. Not only has this given me an insight into my own behavior, but now I’m better prepared to help the individuals on my team thrive and communicate under different levels of pressure on a personalized level. Take note that your behavior preference also impacts how you receive feedback and communicate upward as well and giving this assessment to those either lateral to or above you in the organizational hierarchy can also yield incredibly useful results. Insight gained from this tool crosses so many boundaries, and if others are open to it, they can actually use the results to improve their awareness and regulation.
Speaking of, during our EQ training, we dove into 4 components of EQ – self-awareness, self-regulation, social awareness, and relationship management. For self-awareness, we’re encouraged to be curious and name our emotions (name it to tame it), dig for reasons behind that reaction, search for patterns or triggers to this emotion, and then lean into the discomfort of that to experience growth. In self-regulation, we were asked to breathe using the 4-7-8 method and practice grounding exercises. Essentially, you want to catch yourself when your brain is going offline and not react until you can engage the prefrontal cortex in a healthy and productive (professional) way. You also have to commit to the practice of self-awareness and self-regulation, making it a habit so that you can hold yourself accountable and build trust both within yourself and across your team.
When it comes to social awareness, take time to read the room. Look at body language and recognize if others have the time or space for the information or energy you are bringing, and put yourself in the other person’s shoes. For relationship management, it’s important be open and listen without judgement or assumptions. Build trust by being consistent and constructive with feedback, and most importantly, develop others by being conscious of needs and encouraging buy-in. Whether you are in-tune with yourself or you’re still navigating the self-section of EQ, one of my biggest takeaways from these sessions is to take time and observe how others manage their emotions. Are they bringing stress from home to work with them, or are they bringing their stress from work home? Notice their nonverbal communication or body language, their timing, how they deliver messages, and how they respond to feedback. How do their reactions impact the rest of the team? How does it compare to how you manage your own emotions? Are you self-aware and actively practicing self-regulation in your everyday life, and are you setting an example for them even if they are dysregulating?
I began thinking about how the LOD tools I received in this program can help anyone who is willing to learn navigate difficult situations in daily life, well beyond the workday. It prompted me to reflect on how my behaviors and reactions in both my professional and personal lives are not only interrelated, but consistent. I reached a point a few years ago where emotions and behaviors aligned just right, and while I am human and burnout can manifest in many ways, the regulation has helped me surpass the glass ceilings I had at that time. In an ideal world, leaders should be able to coach others to recognize and regulate so they can exceed all expectations they’ve set for themselves. If you say you want to be a leader and shatter your professional glass ceiling, keep in mind that it will never be handed to you. Simply excelling at the laboratory skills associated with your job is not enough to be a leader as low EQ will unveil itself immediately. As a leader, you need to be able to regulate your emotions and practice social awareness and relationship management. I encourage you to self-reflect and assess your emotions and behavior both at home and at work, in your family life, and in your social circles, under stress and when you find peace. As for relationship management, understanding your reactions and the behaviors of your team members provides insight to the overall team’s wellbeing and how it shapes the culture of the organization. While these trainings are required for new leaders in our health system, I can’t help but feel that most should also be required for all employees. Things like behavior exploration, EQ, feedback, and communication – these are critical soft skills that all employees could benefit from professionally. But it’s more than that – these tools can help you succeed in your personal relationships and overall well-being. With ongoing self-regulation and relationship management, you can break every glass ceiling you hold over yourself. It’s okay if you’re a work in progress; think of it as personal continuous improvement.
-Taryn Waraksa-Deutsch, DHSc, SCT(ASCP)CM, CMIAC, LSSGB, is the Cytopathology Supervisor at Fox Chase Cancer Center, in Philadelphia, Pennsylvania. She earned her master’s degree from Thomas Jefferson University in 2014 and completed her Doctorate of Health Science from Bay Path University in 2023. Her research interests include change management and continuous improvement methodologies in laboratory medicine. She is an ASCP board-certified Specialist in Cytology with an additional certification by the International Academy of Cytology (IAC). She is also a 2020 ASCP 40 Under Forty Honoree. Outside of her work, Taryn is a certified Divemaster. Scuba diving in freshwater caverns is her favorite way to rest her eyes from the microscope.
As we begin a new year, we have two important roads to travel. The first is to see where we went in the year prior. January is a great time when we can sit down and draft our year-end reports. We look back at safety audit scores, calculate our injury and exposure rates, and we compare ourselves to industry standards and measure our success and ability to meet those goals.
The other is the road ahead. We can take what we’ve learned and lay out a path to improve what we saw in the past year(s). So, I’d like to challenge all of you to a commitment to improving lab safety, a New Year’s resolution. But what is a resolution exactly? Is it simply a decision or is there more to the definition of the word? Let’s pause for a second and take a deeper look into the meaning behind this word resolution.
A few synonyms for resolution are the act of resolving (an issue), answering (a question), solving (a problem), or analyzing (a situation). When we make a New Year’s resolution to lose weight, what are we trying to do? We’re trying to resolve the problem of unhealthy eating habits, or to analyze our exercise routine. Maybe you are trying to resolve safety issues in your lab. What can you do? It is easiest to start with the low hanging fruit, of course, but you also need to be on guard that the fruit doesn’t come back. This year I challenge you to reach a little higher by resolving safety culture issues that lead to the return of that low hanging fruit.
When it comes to PPE compliance, the best thing you can do is be there for your staff. What does that mean? It goes beyond just having PPE available for your staff, that is something that is required. To truly be there for your staff with regard to PPE means that you approach staff and talk to them about PPE issues. It also means that you demonstrate or model proper lab attire and act as the gold standard for PPE usage. Your staff will pay attention to your behaviors, and they will mimic your actions. That means when you’re having huddles or leading a meeting in the lab, make sure to don a lab coat. If you have to print something from the computer, don’t forget to reach for that lab coat and gloves – if the equipment is in the lab area, it’s surfaces should be treated as potentially contaminated.
Another great way to show the staff you’re interested in safety is to bring it up often. Make sure you normalize discussions around PPE in your huddles and monthly meetings. There’s a reason why weight loss apps and programs that incorporate community involvement tend to work better than just telling ourselves, that’s it no more chocolate for the rest of the year. We all know that’s not going to work. Neither is putting up a sign in the department that reminds staff to wear PPE or assigning another computer-based learning module around PPE usage. It takes a little more effort to resolve the overarching issue of compliance in order to make a lasting difference.
I don’t know about you, but one thing I’d like to solve is the issue we see in the lab regarding the use of cell phones and other personal electronic devices. What options do we have as lab leaders? Some labs have gone to extremes to ban all cell phones in the lab. There have been success stories in some labs, and these policies have backfired horribly in others. To resolve this problem, we first have to start with what we want to accomplish. Overall, keeping cell phones out of dirty areas is of the greatest importance. We know why it’s a bad idea to introduce this electronic device in the lab area that is contaminated with viruses, bacteria, fungus, and who knows what else. The real question is how – how do we get staff to truly understand the risks of using cell phones in the lab, and how do we keep them from the behavior of wanting to pull their cell phones out inside of a contaminated workspace.
The noted behaviorist and lab biosafety expert, Sean Kaufman, compares the addiction to cell phones and social media to that of nicotine. Years ago in workplaces, five or ten-minute smoke breaks were quite common and occurred every hour or two. Kaufman suggests that it may be appropriate to bring that back, giving staff a cell phone break or an “e-break” instead. At first this practice might seem like it could jeopardize the lab’s productivity. However, when you think about how often staff are stopping work and glancing at their phones, it might actually improve productivity and restore engagement in the work (fewer distractions, fewer mistakes, and less negative impact to laboratory results).
They say it’s true that you catch more flies with honey than with vinegar. In published studies, the use of positive reinforcement has shown greater success when trying to change behavior. Certain forms of positive reinforcement such as verbally praising proper behavior in the moment can work. Some labs have developed a point system where staff can earn points for being “caught” in good safety moments. Staff can then take points which they accumulate and redeem them for prizes such as T-shirts, pens, etc.
But what if positive reinforcement just doesn’t cut it? At times it is necessary to hold staff accountable. I ask you this year to go out and look at your labs and notice how many times you see somebody on their cell phone. Then ask yourself what is an acceptable number of occurrences? One? Ten? If you ask your staff to not use their cell phones and they continue to do so, ask why they’re not following procedures, but more importantly, why they are not listening to your request. Remember, as a lab manager or leader, you are ultimately responsible for safety in the lab. If you turn a blind eye to somebody using their cell phone, chances are they will continue to repeat that behavior. They will continue to use their cell phones until something bad happens. Before you know it, that employee could accidentally report an incorrect result on a patient because they were distracted, or they may become ill because of a pathogen they brought home on the phone. If something like that were to happen, who do you think would be at fault? Is it the employee or the manager who noticed the unsafe behavior and allowed it to continue?
Sure, you can make a resolution or commitment to improve your lab safety. You know there are issues, you want to fix them, and deep down you really know what you have to do. But it’s not easy. They say one of the best ways to ensure that you follow through with a commitment is to let others know of your plan and to have them help hold you accountable. As mentioned before, community-based programs tend to work better than going at change alone. Rather than make a resolution to improve your lab safety, start with making a resolution to discuss safety in your lab more often. Hold others accountable and allow others to hold you accountable as well.
-Jason P. Nagy, PhD, MLS(ASCP)CM is a Lab Safety Coordinator for Sentara Healthcare, a hospital system with laboratories throughout Virginia and North Carolina. He is an experienced Technical Specialist with a background in biotechnology, molecular biology, clinical labs, and most recently, a focus in laboratory safety.
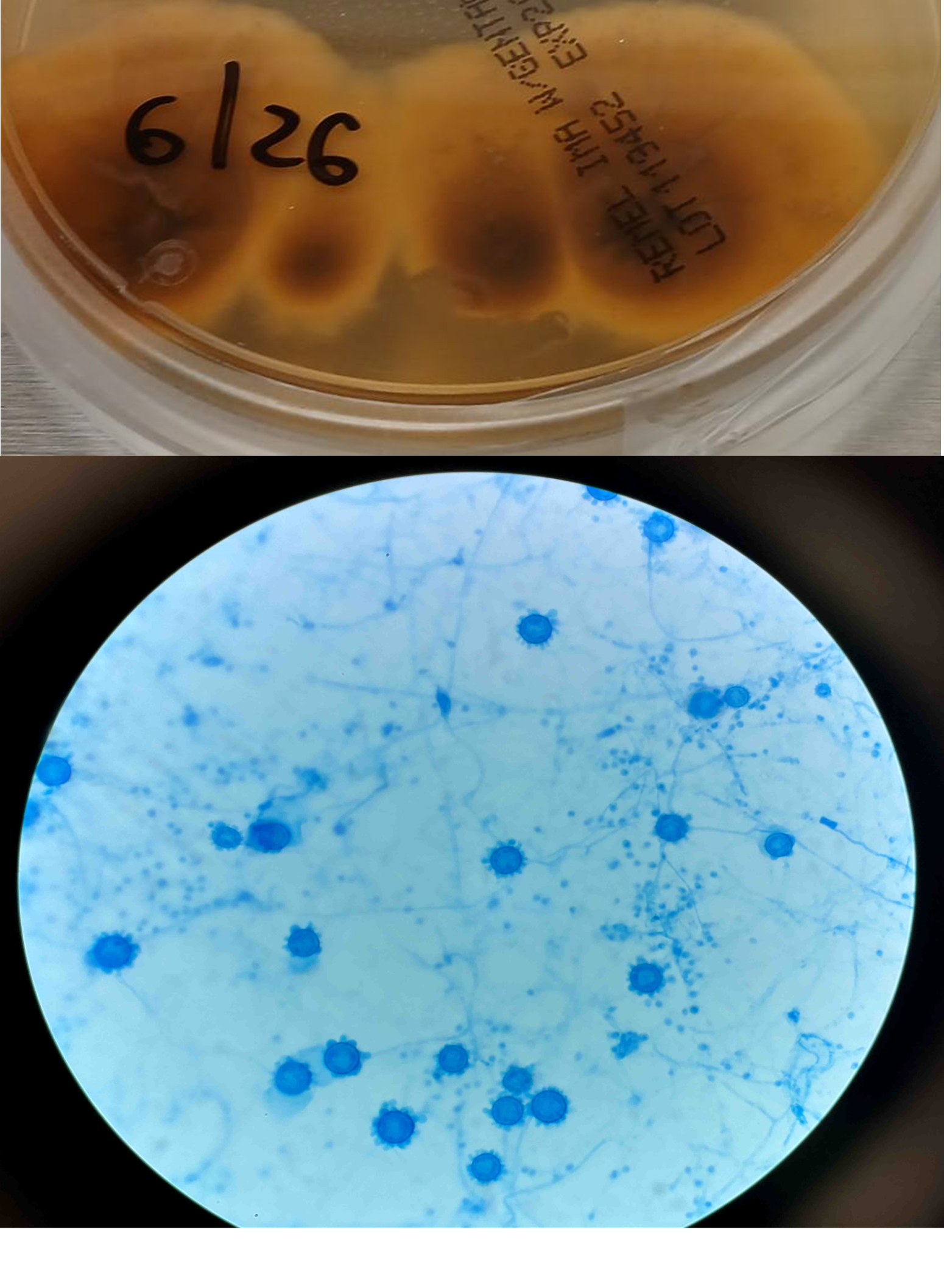
A 41-year-old male was admitted with myasthenia gravis exacerbation and respiratory difficulty. He was diagnosed with myasthenia gravis in 2004, underwent a thymectomy in 2010, and was taking prednisone (20 mg daily). He presented to another hospital one month ago with fever, chest pain, and shortness of breath. He was treated with antibiotics and antifungals for pneumonia, along with 5 days of immunoglobulin for a possible myasthenia gravis crisis, but showed no improvement. After admission, a CT scan revealed diffuse bilateral mixed airspace and ground-glass opacities without pleural effusions, most concerning for multifocal pneumonia. A bronchoalveolar lavage (BAL) with Gomori methenamine silver (GMS) staining showed hyphal and yeast elements. Both BAL and urine Histoplasma antigen tests were positive. He was started on posaconazole (300 mg daily), which led to an improvement in symptoms, and he was subsequently discharged. After 6 weeks of culture, macroscopic and microscopic features of the colony confirms Histoplasma capsulatum (Figure 1).
Figure 1. Macroscopic analysis reveals dark brown color on reverse (top). Lactophenol cotton blue preparation of mold reveals tuberculated, macroconidia (bottom).
Discussion
Histoplasma capsulatum is a dimorphic fungus responsible for histoplasmosis, a respiratory infection. As with other dimorphic molds, this fungus exists as a mold in the environment at 25°C and converts to a yeast form once it infects a host at 37°C. It is primarily endemic in areas with rich soil, particularly in the Ohio and Mississippi River valleys in the United States, but has also been reported in Maryland, Pennsylvania, Delaware, West Virginia, Virginia, and North Carolina. Outside the United States, histoplasmosis incidence is best understood and highest in parts of Mexico and South and Central America and is largely driven by the AIDS pandemic1,2. The fungus thrives in bird and bat guano due to the high nitrogen and organic content; common exposures include bird roosts, chicken houses, caves, and old buildings that are commonly visited by bats. Humans typically become infected through the inhalation of aerosolized spores. Demolition or building construction around these soiled environments can lead to outbreaks.
While most infections are asymptomatic, histoplasmosis can lead to severe illness in immunocompromised individuals. Histoplasma spores are deposited in the lungs, where they convert to the yeast form. These yeast cells are phagocytosed by alveolar macrophages, where they survive and multiply inside the cells. The fungus can then disseminate via the lymphatic and bloodstream systems to other organs 3. The clinical presentation of histoplasmosis varies depending on the host’s immune status and the level of fungal exposure. In most cases, the infection is asymptomatic or manifests as mild flu-like symptoms, including fever, fatigue, and cough. However, in cases of acute pulmonary histoplasmosis, patients may experience pneumonia-like symptoms such as chest pain, cough, and fever. Chronic pulmonary histoplasmosis can mimic tuberculosis, presenting with symptoms such as weight loss, night sweats, and a chronic cough. Pleural thickening and apical cavitary lesions are common. Extrapulmonary manifestations include granulomatous mediastinitis, fibrosing (sclerosing) mediastinitis, and broncholithiasis1. In severe disseminated cases, particularly in immunocompromised individuals, the infection can spread to multiple organs. Adrenal involvement may manifest as adrenal masses, adrenal insufficiency, or electrolyte imbalances. Gastrointestinal involvement is relatively common but rarely produces clinical symptoms4. Skin involvement occurs in up to 15% of cases in studies, more frequently in patients with AIDS. Central nervous system (CNS) involvement occurs in 5-20% of cases5.
Diagnosing histoplasmosis involves clinical suspicion, radiological studies, and laboratory tests. The most common radiographic findings are diffuse reticulonodular pulmonary infiltrates. Cavitations are seen in chronic cavitary pulmonary histoplasmosis, and mediastinal or hilar lymphadenopathy is often present. In immunocompetent individuals, infections may lead to development of granulomas that may or may not be necrotizing.
For laboratory testing, antigen detection in urine or serum using enzyme immunoassays has a sensitivity of 90%6 in disseminated disease and 75%7 in acute pulmonary disease. Antibodies may take 2-6 weeks to appear in circulation and are useful in chronic cases to confirm suspicions of infections but less effective for detecting acute infection and in immunosuppressed patients with a poor immune response.
Culturing Histoplasma from clinical specimens is definitive, but it can take 4-8 weeks due to the slow growth of the fungus. The colonies appear white at young growth, have a cottony, cobweblike-aerial mycelium and can mature into brown or grey color on reverse (Figure 1, top) 8. Microscopic examination of mold colony using lactophenol cotton blue preparations reveals the characteristic large, rounded, tuberculate macroconidia (circular structures with roughened/spiked edges) originating from short, hyaline conidiophores (Figure 1, bottom). Histoplasma capsulatum appears as small (2-5 μm), oval, intracellular yeast cells typically found within macrophages. The yeast may exhibit a clear space which may appear like a capsule, but is actually a retraction artifact due to the processing of the specimen. Staining with GMS or periodic acid-Schiff (PAS) highlights the yeasts9. Molecular methods such as PCR and sequencing have been shown to detect cases of Histoplasma as well.
Treatment varies depending on the severity of disease. Mild cases, particularly those involving acute pulmonary histoplasmosis, often resolve without specific treatment, although antifungal therapy (e.g., itraconazole) is recommended in more severe cases. For chronic or disseminated histoplasmosis, initial treatment with amphotericin B is often followed by long-term itraconazole therapy. The prognosis is favorable for immunocompetent individuals with mild disease, but disseminated histoplasmosis can be fatal in immunocompromised patients if not treated promptly.
References
1. Wheat LJ, Azar MM, Bahr NC, Spec A, Relich RF, Hage C. Histoplasmosis. Infect Dis Clin North Am. 2016 Mar;30(1):207-27. doi: 10.1016/j.idc.2015.10.009. PMID: 26897068.
2. Bahr NC, Antinori S, Wheat LJ, Sarosi GA. Histoplasmosis infections worldwide: thinking outside of the Ohio River valley. Curr Trop Med Rep. 2015 Jun 1;2(2):70-80. doi: 10.1007/s40475-015-0044-0. PMID: 26279969; PMCID: PMC4535725.
4. Sarosi GA, Voth DW, Dahl BA, Doto IL, Tosh FE. Disseminated histoplasmosis: results of long-term follow-up. A center for disease control cooperative mycoses study. Ann Intern Med. 1971 Oct;75(4):511-6. doi: 10.7326/0003-4819-75-4-511. PMID: 5094067.
5. Wheat LJ, Batteiger BE, Sathapatayavongs B. Histoplasma capsulatum infections of the central nervous system. A clinical review. Medicine (Baltimore). 1990 Jul;69(4):244-60. doi: 10.1097/00005792-199007000-00006. PMID: 2197524.
6. Wheat LJ, Kauffman CA. Histoplasmosis. Infect Dis Clin North Am. 2003 Mar;17(1):1-19, vii. doi: 10.1016/s0891-5520(02)00039-9. PMID: 12751258.
8. Azar MM, Hage CA. Laboratory Diagnostics for Histoplasmosis. J Clin Microbiol. 2017 Jun;55(6):1612-1620. doi: 10.1128/JCM.02430-16. Epub 2017 Mar 8. PMID: 28275076; PMCID: PMC5442517.
9. Hage, C. A., Ribes, J. A., Wengenack, N. L., Baddour, L. M., Assi, M., McKinsey, D. S., … & Wheat, L. J. (2010). A multicenter evaluation of tests for diagnosis of histoplasmosis. Clinical Infectious Diseases, 50(4), 508-512.
-Xingbang Zheng, M.D., was born and raised in Hefei, China. He attended the Peking University Health Science Center, where he received his doctorate degree. He then worked as an OB/GYN physician in China, primarily focusing on female infertility and reproductive surgery. His clinical research concentrated on subtle distal fallopian tube abnormalities and their relationship with endometriosis and infertility. In his free time, Xingbang enjoys swimming, visiting museums, and spending time with his family. Xingbang is currently pursuing AP/CP training.
-Rebecca Yee, PhD, D(ABMM), M(ASCP)CM is the Chief of Microbiology, Director of Clinical Microbiology and Molecular Microbiology Laboratory at the George Washington University Hospital. Her interests include bacteriology, antimicrobial resistance, and development of infectious disease diagnostics.
When the Medical Examiner’s office receives a report of someone dead in a bathtub, the reporting party often presumes that the individual must have drowned. Yet deaths in bathtubs can be surprisingly complex. We initially discussed drowning back in June of 2023 but to summarize, drowning is a diagnosis of exclusion. The few signs of drowning at autopsy (pulmonary edema, watery fluid in the stomach and sinuses), are neither sensitive nor specific, so it is critical to exclude other potential causes of death. Particularly when a bathtub (or hot tub) is involved, a methodical step-by-step approach is helpful to avoid jumping to inaccurate conclusions.
The first step for us is to determine if the tub contained water. While this may sound obvious, this critical detail isn’t always reported initially. The first person on scene often reflexively turns off running water and opens the drain – and (understandably) in the heat of the moment, this alteration of the scene might not be documented. Our investigators are experienced at identifying indirect evidence of a wet bathtub, such as a water line, wet clothing, or wet sponges and washcloths.
Simply having water in the tub isn’t sufficient, though. We need to know whether the person’s nose and mouth were beneath the water. Sometimes, the physical dimensions of the tub and the position in which the person was found are incompatible with complete submersion. If the body has been moved, though, this evidence may be lost. Conversely, just because someone’s face is under the water doesn’t prove that they drowned; someone may die suddenly from heart disease while filling the tub, for example, and subsequently the water rises.
As mentioned earlier, there are no pathognomonic signs of drowning at autopsy. This isn’t necessarily a problem in the proper context. Take this example: a person who doesn’t know how to swim (and isn’t wearing a flotation device), falls out of a boat into a natural body of water. The body is recovered a day later. An autopsy reveals no “typical” drowning findings, but no other potential causes of death. In this scenario, drowning is the only remaining reasonable option for a cause of death. But to diagnose drowning in a shallow body of water, we need to explain why the person couldn’t escape the environment. In other words, in a bathtub, why didn’t the decedent just sit up? Sometimes, the reason is the age of the deceased – infants or young toddlers clearly can’t escape a deep bathtub, but even large buckets can be dangerous if they fall head-first (figure 1). Elderly and frail individuals, or those with neuromuscular conditions, may be unable to pull themselves up to a sitting position. Conditions like epilepsy can be dangerous if a seizure occurs while the person is bathing, and intoxication by alcohol, opiates, or other sedative-hypnotic drugs may cause someone to lose consciousness and slip beneath the water.
As you can see, there are a lot of avenues for interrogation investigating a death in a bathtub. That’s why these cases can be excellent examples of forensic casework – the correct answer is only identified following thorough scene investigation, autopsy, toxicology, and a review of the medical history.
Figure 1: This illustration released by the U.S. Consumer Product Safety Commission warns that 5-gallon buckets, in particular, are a drowning hazard for young children.
As the year winds down, laboratory leaders often find themselves balancing the usual workload with holiday schedules, supply ordering, and end-of-year tasks. But amidst the whirlwind of activity, one key area should remain front and center: laboratory safety. Wrapping up the year with a focus on safety isn’t just about meeting compliance standards—it’s an opportunity to reinforce your culture of safety, recognize team achievements, and set a strong foundation for the year ahead.
But how can you end your lab safety year on a high note, ensuring that your lab team feels valued, prepared, and motivated for the challenges to come? There are some methods you can use.
First, before diving into new year initiatives, take a moment to assess the past year. What worked well, and what could be improved? When looking at accident and incident reports, were there any recurring issues? If so, identify root causes and develop targeted interventions. Review inspection findings from internal audits, accreditation agencies, or regulatory bodies. Address any outstanding corrective actions. Verify that all team members have completed their required training, and close any gaps you find. This reflection phase provides valuable insights and ensures you’re closing out the year without leaving loose ends.
Next, take a look at your lab safety policies and procedures for updates. Laboratory safety policies should evolve alongside changes in equipment, workflows, and regulations. Use this time to ensure that Standard Operating Procedures (SOPs) are current and reflect actual practices. Engage your lab team in identifying outdated or unclear safety policies. Check to see if new materials, processes, or technologies introduced new potential risks. If so, update your risk assessments accordingly. By aligning policies with the current state of your laboratory, you demonstrate a commitment to proactive safety management.
Celebrating safety successes at the end of the year is important, too. Recognition is a powerful motivator. Highlighting your team’s achievements in safety reinforces the importance of their efforts. Be sure to recognize individuals or teams for exemplary safety practices, such as reporting near-misses or suggesting improvements. If your lab reduced incidents or improved compliance, share those results during team meetings or in a newsletter. Did someone design a clever safety poster or come up with an innovative solution to a hazard? Acknowledge their contributions as well. Celebrating successes boosts morale, raises safety awareness, and encourages ongoing engagement in safety programs.
The end of the year is also a perfect time to address outstanding safety tasks that may have been sidelined. Conduct a thorough chemical inventory, disposing of expired or unnecessary chemicals according to your facility’s guidelines. Verify that eyewash stations, safety showers, fire extinguishers, and spill kits are in working order and properly stocked. Replace faded or damaged labels and signs to ensure they’re clear and compliant. Clearing these tasks from your list not only improves safety but also sets a standard of organization for the upcoming year.
Remember, lab safety is a shared responsibility, and the end of the year is an excellent time to foster a sense of ownership among your team. Hold a safety roundtable by inviting team members to discuss safety concerns and suggest improvements. Gather anonymous feedback on your lab’s safety culture, policies, and practices. Involve your team in setting safety goals for the upcoming year. This could include launching new initiatives, revising training programs, or addressing specific hazards. Engagement fosters collaboration and ensures that everyone has a voice in creating a safe work environment.
A successful safety program doesn’t stop at the calendar year. Use this time to lay the groundwork for a strong start in the new year. Schedule annual training by planning sessions to refresh key safety concepts, such as bloodborne pathogens, chemical hygiene, or emergency procedures. Setmeasurable goals like reducing near-misses by 10% or achieving 100% compliance with safety training. Update all emergency contacts to ensure they are current and accessible to your team. A little preparation now can save time and prevent confusion down the road.
Finally, make it a habit to end the year with gratitude. Laboratory work is demanding, and your team’s commitment to safety deserves acknowledgment. Start with written thank-you notes. A personal message to each team member can go a long way in showing your appreciation. Celebrate the year’s achievements in a relaxed setting, and remind your team how their efforts contribute to the lab’s success and safety culture. When people feel valued, they’re more likely to stay engaged and committed to safety.
Ending the lab safety year on a high note requires focus, organization, and a genuine commitment to your team’s well-being. By reflecting on the past, addressing current needs, and preparing for the future, you can create an environment where safety isn’t just a requirement—it’s a shared value. Take the time to celebrate successes, involve your team in decision-making, and express gratitude for their efforts. These steps not only close out the year on a positive note but also build momentum for continued safety excellence. Here’s to a safe and successful new year in the laboratory!
–Dan Scungio, MT(ASCP), SLS, CQA (ASQ) has over 25 years experience as a certified medical technologist. Today he is the Laboratory Safety Officer for Sentara Healthcare, a system of seven hospitals and over 20 laboratories and draw sites in the Tidewater area of Virginia. He is also known as Dan the Lab Safety Man, a lab safety consultant, educator, and trainer.
Prior to my arrival in the laboratory in April 2020, we had a written procedure to repeat any cerebrospinal fluid (CSF) HSV-1 or -2 positives with a CT value >38. Upon arriving I was asked by our molecular supervisor if I wanted to keep this rule. Not having specific experience with this instrument and assay and knowing this was an FDA approved direct sample to answer assay, I asked “what does the package insert (PI) say?” When I reviewed the PI and further discussed with our supervisor, we decided this was not in the PI and even though we could review the curves, we should treat results from this assay like a “black box” direct sample to answer assay. The black box being referred to here, is a sample to answer instrument in which the cycle threshold (Ct) values are not visible to the user.
We updated the procedure to report the results that the assay produced and removed repeating any results that were not invalids or errors.1 This decision was based on several factors. 1. Repeating high CT value results are likely testing a bioburden that is near the limit of detection (LOD) and could repeat as positive but repeating as negative does not invalidate the first positive result. 2. The picture could get muddied by changing a positive to a negative based on a repeat result. Calling the clinician with conflicting results was more confusing to the provider than the provider reaching out to say the positive did not make sense and then sharing the high CT value. 3. We had no basis, ie validations to demonstrate that the higher CT values were not reliable/accurate. College of American Pathologists checklist item, CAPMIC.64975 Modified Cut-Off Phase II “If the laboratory has modified the manufacturer’s cut off-value for a positive result, the new cut-off value has been validated.”2
Technologist vignette
While this repeat was removed from policy, my team remained vigilant of when those high CT values would report out.
One weekend out of previous habit, a CSF HSV-1 PCR positive with high CT 37.9 was repeated and it was negative. The weekend consult I received was “Should I report the original positive or the repeat negative? I’m reviewing the CSF cell counts and seems like the positive could be real?” After review and discussion, this repeat was a result of still being nervous about the higher CT value and repeating even though it was no longer in the policy. This also ended up being a missed opportunity for contamination check because the repeat test was performed after disinfecting the instrument. Environmental swabs and QC were not run before that second test. Was it potential contamination or was it just a low level positive? The positive was reported and the clinician did not have additional questions.
Pediatric Infectious Disease Provider Vignette
One week later, I was alerted to a pediatric patient with CSF HSV-1 PCR positive result with a high CT value (39.4) but the Enterovirus (run on a separate instrument) was also positive at a CT value of 27.4. Both positive results were posted in the EMR and I was consulted by the pediatric ID provider requesting consultation on validity of results. I shared that seeing this very high CT value paired with the Enterovirus makes me consider the possibility of a false positive. With the predominance of HSV in the community a false positive can be introduced at any point including specimen collection. To alleviate laboratory contamination concerns, environmental testing was performed with no indication of contamination. I assured the physician that there is no additional manipulation of the specimen once in the lab other than testing setup. I shared repeat testing will not be helpful. Repeat testing was not requested on the original specimen. Our ID provider was on board with ruling it a contaminant or insignificant to the diagnosis after a repeat CSF was collected and tested negative.
Phone a Friend Vignette
This prompted me to have a conversation with a colleague about these results. I learned that at their institution they incorporated a comment for any CT value >=35. “This specimen yielded a low positive result, which may not be reproducible and should be interpreted in the context of the patient’s clinical presentation.” We opted to adopt this statement at our institution to aid us in conversations with providers. At the LOD, it will be challenging to confirm the results as there really isn’t a way to do it without uncertainty surrounding the results.
Adult Hospitalist Investigation Vignette
Almost one year later I received an email forwarded to me by our medical director from a physician that was sharing concern over our HSV-2 test results.
“Over my ten-year career, I have encountered only a handful of HSV-2 cases. Interestingly, in the last two months alone, I’ve observed three HSV-2 cases in the same unit!!
In the first two cases, the CSF profiles were remarkably benign, making an HSV-2 positive result highly unexpected. Despite this, both patients tested positive for HSV-2 (4/18 and 5/17) and were treated accordingly.
The third case involved a patient diagnosed with cryptococcal meningitis, which accounted for the CSF findings. However, the HSV-2 test also returned positive (6/5), which was surprising given the extremely low likelihood of concurrent infection with cryptococcal meningitis and HSV-2. After initiating treatment with amphotericin, the patient began to improve. When the HSV-2 results came back, we started acyclovir. However, two additional samples taken within 24 hours both tested negative. It is well-documented that true HSV-2 infections typically maintain positive results for an extended period, even with appropriate treatment.
Given these observations, I am concerned that we may be dealing with laboratory errors, leading to the overtreatment of patients based on false positives.
Could you please investigate these cases further?”
My response as Infectious Diagnostics Director
Good afternoon concerned provider. We conducted a review of our results, quality control (QC), and have corresponded with the manufacturer. We have had no QC contamination monitoring concerns. The result PCR curves did not demonstrate anything questionable (the result appears to be a real positive without abnormal signaling). All three of the patients you shared had results that were at the lower end of detection. While, the manufacturer, did not recommend to do this, we do add a comment for CT values >35 (towards the lower LOD) that the specimen yielded a low positive result which may not be reproducible and should be interpreted in conjunction with patient presentation. Snip from chart below. Any tests at the lower limit of detection are challenging because when virus is truly there it will likely not repeat. We will be monitoring this closely and continue to report trends to the manufacturer and continue to monitor our internal quality control per protocol and review the need for added quality control monitoring with other users.
Low pre-test probability does increase the risk of false positives, however, this test has been cited in the literature to have high accuracy, but this may be skewed toward truly positive cases. The test we use is a targeted assay as described in the attached article3. We are in process of pushing forward an investigation with the manufacturer and I will share if there are further findings.
If you’d like to chat to better understand any of this you can reach me via secured messaging or my cell.
The follow up discussions
The provider did want to have further conversation because the HSV-2 positive result simply did not make clinical sense to him. After discussing and not being satisfied by the possibility of low level HSV-2 in the CSF, I offered a review by our infectious diseases providers. Infectious disease was involved in all three cases during their encounter.
For the first case, ID provider shared that CSF does not correlate with HSV but clinically with mental status change there was no other clear source of encephalopathy. For the second case, the CSF cell count did not correlate with HSV but since patient had a prior history of transplant and was on immunomodulator there was a high risk for mollaret meningitis, rare form of meningitis that is recurrent, aseptic, mild, and self-limiting, HSV-2 being the most commonly associated agent. At the time of initial treatment, patient had mental status change. The third case was deemed to be either a contaminant or false positive by the ID provider consulted. This assessment was made since his abnormal CSF was explained by patient’s Cryptococcal meningitis and the repeat HSV was negative without treatment.
Only 1 of the 3 was fully confirmed by ID to be considered a contaminant. Given the prevalence of HSV-1 and HSV-2 at various body sites among the healthy population, human contamination is a necessary consideration for providers when the results do not make clinical sense.
What we did and what we changed
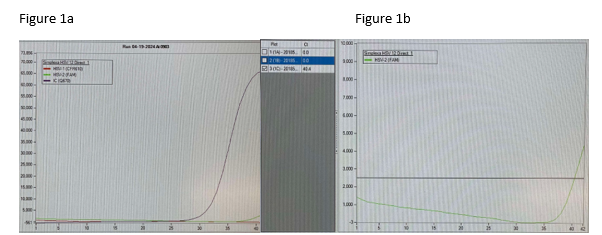
Following multiple emails and conversations with the manufacturer scientific liaison in which we reviewed each curve that was questioned, we did not change our response to the provider. However, we gained valuable insight into what an instrument contaminant would look like and we simply did not see any of that. We saw clean curves that came up at later cycles.
Figure 1 a. Amplification curve of one of the patients in question demonstrating a much earlier signal detection of the internal control compared to HSV-2. b. Close up of HSV-2 signal amplification alone demonstrating late rise without any skips, blips, or other issues that would suggest direct instrument contamination.
We learned of the recommended high touch surface areas to test for environmental testing and the use of a blue plate to minimize contact with the loading disc for testing.
We will likely continue to see high CT values, get questions about potential false positives (some of these stories are patient driven, even when the patient has a previous history and consistent CSF profile), and continue to have in depth conversations with our providers.
What would you do? Better yet – what do our physicians do then? In our case our provider was not comfortable with the response and requested ID review.
What do you do? Please share as we are constantly learning from each other.
References
Diasorin Molecular SimplexaTM HSV 1&2 Direct package insert for both Cerebrospinal fluid and cutaneous and mucocutaneous lesion swabs.
CAP Microbiology Checklist 2023
Gaensbauer JT, Fernholz EC, Hiskey LM, Binnicker MJ, and Campioli CC. Comparison of two assays to diagnose herpes simplex virus in patients with central nervous system infections. Jour Clin Vir. 166(2023) 105528. DOI: 10.1016/j.jcv.2023.105528
-Kimberly Mckean, MLS(ASCP)
-Frances Valencia-Shelton, PhD, D(ABMM), SM(ASCP)CM is the Clinical Infectious Diagnostics Director for the Baptist Health System in Jacksonville, FL. She is actively engaged in the Jacksonville Area Microbiology Society and the American Society for Microbiology. Her interests include defining and utilizing clinical best-practice for testing and reporting. She is equally interested in learning with and educating others in the field of clinical microbiology.