Case History
A 23 year old man presented to the hospital with recurrent fever up to 103F with associated nausea and vomiting, epistaxis, watery diarrhea, dyspnea, and decreased appetite for several days. Blood cultures from admission were positive for MSSA and a stool PCR was positive for Vibrio species. He was admitted and treated for sepsis. His CBC demonstrated a marked pancytopenia ( WBC count 0.6 K/μL) and the hematopathology team was consulted to review the peripheral blood film.

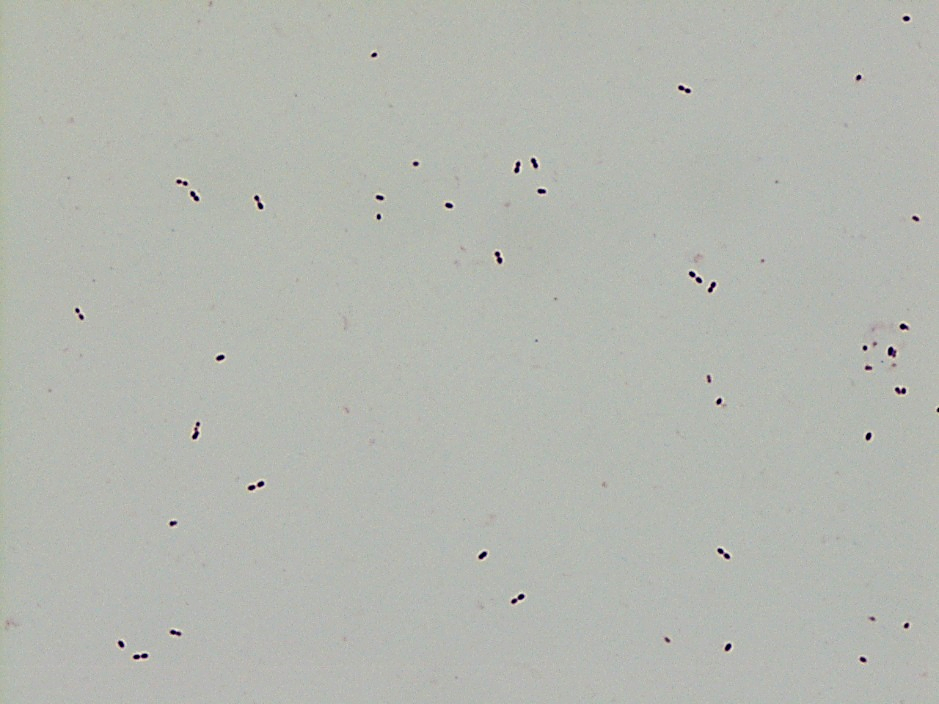
Review of the peripheral blood confirmed a markedly pancytopenic picture with virtually no leukocytes in the region of best RBC “spread” (Image 1A). In the periphery of the smear (1B and C) clusters of leukocytes were noted where left-shifted granulocytes were seen. Many demonstrated nuclear irregularity and abnormal granulation (B) and some showed the presence of numerous Auer rods (Image 1C, arrows).
The presence of abnormally granulated immature neutrophilic precursors, and cells with numerous Auer rods was morphologically compatible with acute promyelocytic leukemia (APL) and a rush preliminary diagnosis was rendered. The patient was started on ATRA therapy and FISH for PML-RARA was expedited.
Discussion
Acute promyelocyticleukemia (APL) is characterized as an acute myeloid leukemia in which promyelocytes with the PML-RARA fusion predominate. The PML-RARA fusion is the result of a balanced translocation between chromosomes 15 and 17, designated ast (15;17)(q24.1;q21.2). The promyelocyte progenitor cell is the cell of origin of APL. APL occurs most frequently in middle aged individuals, but can occur at any age.
The first account of APL was originally discussed in the late 1950s in which L. K.Hillestad, a hematologist from Norway, described a disorder as “a white blood cell picture dominated by promyelocytes and severe bleeding caused mainly by fibrinolysis.” The gene fusion was elucidated in the late 1970s at the University of Chicago demonstrating the balanced translocation between chromosomes 15 and 17. Cure rates at that time were still very low, until in the mid 1980s when researchers in China demonstrated the use of all-trans retinoic acid causing complete remission in APL patients.
Two distinct subtypes of APL exist: hypergranular (typical) or microgranular. The hypergranular variant is filled with large Auer rods and with dense cytoplasmic granules that can obstruct the nucleus. In contrast, the microgranular variant has a scantiness of cytoplasmic granules or small azurophilic granules.
The immunophenotype for APL is quite distinct and characterized by low or absent expression of CD34 and HLA-DR (in keeping with the cellular differentiation from blast to promyelocyte). APL cells are positive CD33 and CD13 with most cases showing expression of CD117 (sometimes weak). APL cells are usually negative for CD15, CD65, CD11a, CD11b, and CD18. The microgranular variant may display positive staining for CD34 and CD2. For both variants, IHC with antibodies to the PML gene demonstrates a nuclear multi granular pattern with nucleolar exclusion, a finding that is unique to APL and not seen in AML or normal promyelocyte morphology.
The main clinical symptom of APL is hemorrhagic, including gingival bleeding and ecchymosis but can progress to disseminated intravascular coagulopathy (DIC). Other symptoms of APL include those related to pancytopenia, including weakness, fatigue, and infections.
The prognosis for APL is considered to be excellent. Tretinoin (ATRA) interacts with the PML-RARA fusion product allowing for maturation and differentiation to occur along the granulocytic lineage, eliminating the promyelocyte population. Combination therapy with tretinoin and arsenic trioxide has become the gold standard of care leading to excellent remission rates.
References
- Kakizuka,A., et al. “Chromosomal translocation t (15; 17) in human acutepromyelocytic leukemia fuses RARα with a novel putative transcription factor,PML.” Cell 66.4 (1991): 663-674.
- Lo-Coco,Francesco, and Laura Cicconi. “History of acute promyelocytic leukemia: atale of endless revolution.” Mediterranean journal of hematologyand infectious diseases3.1 (2011).
- Rowley,JanetD, HarveyM Golomb, and Charlotte Dougherty. “15/17 translocation, aconsistent chromosomal change in acute promyelocytic leukaemia.” TheLancet 309.8010 (1977): 549-550.
- Swerdlow,Steven H. WHO Classification of Tumours of Haematopoietic and LymphoidTissues. International Agency for Research on Cancer, 2017.

-Christopher Felicelli is an M3 at Loyola University Chicago Stritch School of Medicine. Follow Chris on Twitter at @ChrisFelicelli

-Kamran M. Mirza, MD PhD is an Assistant Professor of Pathology and Medical Director of Molecular Pathology at Loyola University Medical Center. He was a top 5 honoree in ASCP’s Forty Under 40 2017. Follow Dr. Mirza on twitter @kmirza.