This post is going to travel slightly off the beaten path by presenting a comparative analysis of my two favorite things – scuba diving and laboratory medicine. What in the world, you say? How can you possibly liken a recreational activity to an entire field of study? Easy. You just have to have a passion for both! I’ve been swimming and snorkeling since I was a wee little kid, always fascinated with being able to free dive down to the bottom of a body of water in one breath (you can blame my mermaid encounter for that). Around that same time, my parents gave me first microscope. The obsession with water was real, always examining wet mounts of everything that could fit between the slide and the coverslip. All throughout my grade school and undergrad years, I flourished in any science course that featured a microscope, finally acknowledging the fact that I had a knack for cells and tissues under the light microscope. During each summer break, I’d travel down to Florida with my parents and embrace the hidden gems of freshwater springs. It wasn’t until my junior year of college where I took a histology course during my summer break that I realized I wanted to make a career of this. During that summer, I began to research scuba diving; however, it wasn’t until after grad school and establishing a career as a cytologist that I finally committed to the idea. 10 years after graduating from a cytology program, I became the cytology supervisor at a renowned cancer center, and six years after my initial scuba diving certification, I became a dive professional, earning the title of divemaster.
Now, you should know that I like to dive deep into things – be it 112 feet down into a dark cavern or fully immersing myself in my work. Here, we’re going to dive into both scuba and the laboratory.
First and foremost, in both fields – diving and laboratory medicine, safety is paramount. There are rules and mechanisms in place to ensure that no one gets hurt. Policies and procedures are designed to keep you, your peers, and your “customers” (patients and students/certified divers) safe. Failure to follow a procedure can jeopardize lives. I’m certain you understand where I’m going with this. There are inherent risks in both fields, and we try to error-proof as best as we can. In diving, safety checks include checking you and your dive buddy’s equipment and configuration to make sure that gas cylinders are turned on and filled to an appropriate pressure, all hoses are connected and work as designed, releases and weights are secured, and that the divers have everything they need to perform the dive. Fortunately, in the laboratory, automation and barcoding has relieved the human eye of some of the burden of solely checking every patient detail, every order, and every equipment’s function. However, laboratory professionals must do their due diligence to double check. No piece of equipment is perfect, but following a procedure designed to avoid errors is the first step in verifying that the correct test has been ordered and the correct results will be delivered to the correct patient.
If you thought that preventative maintenance was necessary for lab equipment, scuba gear is the same way. Unlike a laboratory inspection, diving, for the most part, does not require evidence of gear maintenance to be “compliant” and therefore fit for use. The exception is visual inspection and hydrostatic testing of gas cylinders, but this is more a Department of Transportation rule. With that said, one of the top three reasons for dive accidents is equipment failure due lack of routine maintenance. That’s a risk I’m not willing to take. As much as I wish I had gills, I’m limited to the human form and the environment we are designed to live in. So whether it’s your own life or a family member’s test result, make sure that the relevant equipment is maintained per the manufacturer’s instructions or a more stringent policy or regulation.
One of the many things I practice (while still ensuring a lean mindset) is redundancy. In diving, redundancy comes in the form of equipment. At a minimum, you should have two computers, two flashlights, two air sources, two regulators, two masks, two cutting implements, two people (buddy team), etc. If something goes wrong during a dive, there is a backup that can get you out of jam so that you can safely continue, or if necessary (and more likely the case), end that dive and formulate a plan for future dives. Think of needing to end the dive as a downtime procedure. In the laboratory, redundancy is having two centrifuges, two staining machines, two accessioning workstations, two analyzers (if that’s even in the budget), and other alternative, yet necessary procedures in the event of an electronic or equipment failure. The concept of redundancy is to maintain operations AND safety if something goes wrong.
Lastly, and something that I end my staff huddles or one-on-ones with is personal well-being. In diving, we disclose that it is okay to call a dive at any point for any reason, and a reason does not have to be provided. If you deem yourself physically, emotionally, or mentally unfit for a dive on any given day, call it. Do not muster through and pretend everything will be okay. That’s when divers neglect equipment checks, or fail to watch their gas consumption, depth limits, or time spent underwater. That’s when accidents happen. When you cannot be present for a dive, you cannot be present for your buddy or your students or divers that you are leading. In laboratory medicine, if you are not well, take the day off. I know we are dealing with staffing shortages across the country, I know that it might shift additional work onto a colleague, but you cannot risk jeopardizing patient care because you neglected yourself. Putting on a brave face for others in a world where it’s encouraged to be vulnerable is detrimental to more than just yourself, it’s detrimental to your passions. So take care of yourself and your (buddy) teams so that you can keep your passions safe and productive.
P.S. Sometimes diving and cytology overlap under the microscope, too!
-Taryn Waraksa-Deutsch, DHSc, SCT(ASCP)CM, CMIAC, LSSGB, is the Cytopathology Supervisor at Fox Chase Cancer Center, in Philadelphia, Pennsylvania. She earned her master’s degree from Thomas Jefferson University in 2014 and completed her Doctorate of Health Science from Bay Path University in 2023. Her research interests include change management and continuous improvement methodologies in laboratory medicine. She is an ASCP board-certified Specialist in Cytology with an additional certification by the International Academy of Cytology (IAC). She is also a 2020 ASCP 40 Under Forty Honoree. Outside of her work, Taryn is a certified Divemaster. Scuba diving in freshwater caverns is her favorite way to rest her eyes from the microscope.
A 67 year old female presented to the emergency department with worsening chest pain and shortness of breath for several weeks. Her medical history was notable for diffuse large B cell lymphoma, for which she had started treatment with rituximab and tafastimab-cxix. Her complete blood count revealed severe leukopenia. A chest computed tomography scan showed focal consolidation in the right lower lobe, and broad-spectrum antibiotics and Filgrastim were started to treat her pneumonia. However, her symptoms did not improve. She underwent a bronchoalveolar lavage, which was sent to microbiology for culture. The initial Gram stain of the specimen was unrevealing. Twelve days later, a pathogen was isolated from cultures with acid-fast media and fungal media. On a Sheep’s blood agar plate, white, chalky colonies appeared. A Gram stain of the isolate showed gram-positive organisms growing as branching, beaded filaments. The organisms were further highlighted on a partial acid-fast stain. MALDI-TOF identified the organism as Nocardia farcinica. The patient was started on trimethoprim/sulfamethoxazole and imipenem, and was discharged following clinical improvement.
Images of Sheep’s blood agar plate showing white, dry colonies (left) and Gram stain highlighting beaded, branching filamentous bacteria after culture in MGIT broth (right).
Discussion
Once mistakenly classified as a fungus due to its filamentous, branching morphology, Nocardia spp. are actually gram-positive, aerobic bacteria that belong to the order Cornybacteriales1. Nocardia are normal inhabitants of the soil, where they digest decaying plant matter. However, they are a cause of human disease in susceptible hosts when they are inhaled or enter via the skin2,3 . The major risk factor for infection is an immunocompromised state, particularly defects in cell-mediated immunity. As such, patients with AIDS or lymphoma, those receiving allogeneic organ transplants, and those taking immunosuppressive drugs including high-dose steroids either are more susceptible to infection or suffer more severe disease2,4. Among persons with intact immune defenses, underlying lung diseases such as chronic obstructive pulmonary disease or cystic fibrosis, predispose to infection1.
The most common clinical manifestation of Nocardiosis, particularly in immunocompromised hosts, is a subacute to chronic pneumonia that can wax and wane, and abscesses are a characteristic pathologic feature4. Nocardia can spread via the blood to affect virtually all other organs, with the brain, eyes, and joints being common sites of dissemination1,5,6. Skin infections, more common in immunocompetent persons, include cellulitis and a lymphocutaneous disease that mimics Sporotrichosis4,5. Specifically for Nocardia farcinica, this species have been shown to be involved in disseminated diseases, with most patients being immunocompromised, but in immunocompetent individuals, cutaneous infections have been reported.
Often the first diagnostic clue comes from visualization of gram-positive beaded, thin, branching organisms on direct smears of specimens7. Of note, while the beaded, branching morphology is characteristic, this appearance is sensitive to several culture conditions including media and temperature. Given that Nocardia does not stain fully on Gram stain, it is recommended that a modified acid-fast stain be done as a reflex stain to confirm the suspicions. Organisms, aside from true Mycobacteria, that contain mycolic acids on the cell wall include Nocardia, Gordonia, Dietzia, Rhodococcus, Segmiliparus, Tsukamurella, and Williamsia. However, in some cases, not all organisms can be seen on the direct smear, which warrants cultures to rule out infection but Nocardia spp. can be difficult to isolate in culture either due to low numbers in the initial specimen, or overgrowth by contaminating bacteria3. They grow slowly, usually over the course of weeks3,4. Nocardia can be isolated from most media used to culture bacteria, fungus and mycobacteria3,7. Colonies often appear white and dry but appearance of velvety, powdery, wrinkled, or being heaped are also not uncommon. On the reverse of the plate, the colors may vary and can be brown, tan, pink, orange, red, purple, gray, yellow, peach, or white. The partial acid-fastness of Nocardia spp. is an important corroborating piece of evidence7. Molecular and proteomic methods, including 16S rRNA sequencing and MALDI-TOF allow for definitive confirmation1,7.
A diagnosis of Nocardiosis aids in the selection of appropriate antibiotics and may raise suspicion for disseminated disease, such as brain abscesses. Trimethoprim/sulfamethoxazole remains the antibiotic of choice, and patients usually respond within weeks4. Adding another potent antibiotic such as imipenem helps treat more severe or disseminated disease6. Studies done in-vitro have shown that isolates with resistance to Trimethoprim/sulfamethoxazole are typically susceptible to carbapenems.
References
1. Traxler RM, Bell ME, Lasker B, Headd B, Shieh WJ, McQuiston JR. Updated review on Nocardia species: 2006-2021. Clin Microbiol Rev. 2022;35(4).
2. Steinbrink J, Leavens J, Kauffman CA, Miceli MH. Manifestations and outcomes of nocardia infections. Medicine (Baltimore). 2018;97(40).
-Stevephen Hung MD, PhD is currently a PGY-4 resident at George Washington University Hospital. He graduated from Case Western Reserve University School of Medicine in Cleveland, OH. His academic interests include transfusion medicine, informatics and molecular pathology.
-Rebecca Yee, PhD, D(ABMM), M(ASCP)CM is the Chief of Microbiology, Director of Clinical Microbiology and Molecular Microbiology Laboratory at the George Washington University Hospital. Her interests include bacteriology, antimicrobial resistance, and development of infectious disease diagnostics.
One of the relatively unique components of forensic pathology is the experience of testifying in court. While it isn’t something we do on a daily basis (or even weekly), being well-prepared is an important professional responsibility. After all, another person’s life and freedom can be in the balance.
The most common situation in which we’re called to testify are homicides. Usually these involve gunshot wounds, but they may also involve blunt force, sharp force, or even caretaker neglect. Yet even deaths ruled ‘accidental’ in manner may end up in a court of law – for example, motor vehicle accidents. Occasionally, if there are concerns about the quality of medical care or negligence, we end up involved in civil lawsuits for wrongful death or medical malpractice.
Forensic pathologists are considered “expert witnesses” when they give testimony. The opposite of “expert witness” is a “fact witness.” Fact witnesses are only allowed to testify to their own personal experience – what they’ve seen or heard directly. In contrast, expert witnesses are able to interpret evidence (e.g. autopsy findings) and offer opinions. In order to be allowed to testify as an expert, one’s qualifications must first be recognized and approved by the court. As such, the initial questions we answer are fairly mundane and directed toward eliciting our training history, board certifications, and licensures.
In TV courtroom dramas, testifying is a fraught experience – the question-and-answer sessions are witty, emotionally charged, and often end with some unexpected bombshell. As usual, though, real life fails to be quite as entertaining. My job as a forensic pathologist is to remain emotionally neutral and do the best job I can explaining my findings in a clear and concise manner. The questions, from both prosecution and defense attorneys, are usually straightforward and fair – focused on the autopsy, and what can or cannot be concluded from them. Most importantly, I don’t have any professional stake in the outcome of a trial. The determination of the defendant’s guilt or innocence most often depends on investigative findings beyond those identified at autopsy. Take the example of gunshot wounds – while I can enumerate and describe the individual wounds, I don’t have any information which might identify the person holding the gun. It’s a common misconception that the forensic pathologist is on the “side” of the prosecution – probably because we’re almost always called to testify by the prosecution. But if our ruling doesn’t support the presence of foul play or trauma, any criminal charges fizzle out long before a trial. The evidence I present may not be completely favorable to the prosecution’s version of events, either, which is why it’s so important the defense attorney has the opportunity to bring this information to light.
There are occasions where the autopsy findings are more critical – perhaps in distinguishing a homicide from a suicide or accident. In these cases, an opposing expert witness may be hired by the defense team to critique my report or offer a contrasting opinion. It’s crucial to not take this development personally. The legal system is adversarial by nature, and those charged with a crime are entitled to the best defense possible. Sometimes that requires obtaining another opinion, much like any living patient can seek a second opinion for a serious diagnosis. Maintaining this neutral mentality helps one to stay focused, even in the face of questions which might feel inflammatory or misdirected.
The first time in court can feel very intimidating, with two tables full of lawyers, a judge, and at least twelve jurors paying close attention. For any witness, remembering that you’re only there to speak to the truth might help to calm any nerves. Maintaining a neutral mindset helps keep the focus where it should be – on the science.
-Alison Krywanczyk, MD, FASCP, is currently a Deputy Medical Examiner at the Cuyahoga County Medical Examiner’s Office.
Disseminated intravascular coagulation (DIC) is a serious process in which the proteins responsible for clot formation become overactive. It is vital to note that DIC is not a disease in itself, rather it is always secondary to an underlying disorder, which causes over-activation of the coagulation system.1 This over-activation results in activation of coagulation that consumes platelets, clotting factors, and causes microvascular fibrin thrombi, which can lead to organ failure secondary to tissue ischemia.2 The brain, kidneys, liver, and lungs are the most prone to tissue damage in DIC.
Some symptoms of DIC may include bleeding (from multiple sites), blood clots, bruising, hypotension, shortness of breath, fever, confusion, memory loss, or behavior change.3 Patients always present with symptoms of other disorders frequently associated with DIC such as sepsis, trauma, liver disease, obstetric complications, and malignancy to name a few. In most cases, coagulation does not result in clinical complications, and is not evident until the consumption of platelets and factors becomes overwhelming.1 Once there is overwhelming consumption, it becomes visible through prolonged clotting tests and increasing thrombocytopenia.1 Therefore, timely recognition is crucial in recognizing DIC.
A major challenge of both prognosis and management, of DIC is that it is not a single disease entity. Prognosis largely depends on the treatment of the underlying condition, as the condition is what drives the activation of the coagulation cascade. Treatment is generally supportive, and entails supporting various organs. Support may include the use of ventilators, hemodynamic support, or transfusion of blood components. As well, algorithms have been developed to help guide the management of patients with DIC.
Case presentation
A 31-year-old female presented to the emergency department with complaints of bleeding gums, bruising all over her body, and recurrent nosebleeds for longer than a week. The patient has a history of Acute Promyelocytic Leukemia (APL/AML-M3), and has had symptoms of gingival bleeding, widespread ecchymosis, and recurrent epistaxis for longer than a week. Past fluorescence in situ hybridization (FISH) of the patient’s bone marrow aspirate revealed a t(15;17) translocation. Bone marrow studies also showed blast cells with granulation, dysmyelopoiesis, and 60% promyelocytes with the presence of Auer rods. Immunophenotyping was negative for expression of CD34, and positive for expression of CD13 and CD33. Cytochemical staining tests were positive with myeloperoxidase (MPO), Sudan Black B (SBB), and specific esterase (SE), and negative with non-specific esterase (NSE). As well, reverse transcriptase-polymerase chain reaction (PCR) confirmed the PML-RAR(alpha) fusion gene, consistent with a diagnosis of AML-M3.
The patient has undergone one past treatment of retinoic acid, as well as traditional chemotherapy. It should be noted that some studies indicate a higher incidence of DIC in acute leukemia patients during remission induction with chemotherapy.1 The patient has no family history of the disorder/condition. There are no occupational or social implications/factors, but it is associated with a specific genetic mutation. The patient has no known drug allergies.
Vital signs were normal upon arrival. Upon physical examination, the patient appeared pale and lethargic, displayed widespread ecchymosis, and swollen gums.
Laboratory tests revealed a decreased hemoglobin concentration of 6.0 g/dL (normal, 12.0-16.0 g/dL), and an increased white blood cell (WBC) count of 19.5 x 109/L (normal, 4.5-11.0 x 109/L). The WBC differential showed 91% promyelocytes with heavy granulation, 3% myelocytes, 4% metamyelocytes, and 2% neutrophils. Platelet count was decreased at 72,000/uL (normal, 150,000-450,000/uL). Prothrombin time (PT) was prolonged at 22 seconds (normal, 10-13 seconds) and activated partial thromboplastin time (aPTT) was prolonged at 54 seconds (normal, 23-36 seconds). D-dimer concentration showed an increase at 12 ug/mL (normal, <0.5 ug/mL), and a decreased fibrinogen concentration of 60 mg/dL (normal, 200-400 mg/dL). Plasma levels of plasminogen were 65% (normal, 80-120%), and plasma levels for a2-antiplasmin were 46% (normal, 80-100%). These findings are consistent with DIC with marked hyperfibrinolysis, as correlated by low levels of a2-antiplasmin and plasminogen. The patient was diagnosed as having symptoms associated with DIC secondary to AML-M3.
The treatment for this patient included avoidance of all invasive procedures including biopsies or intravenous (IV) line placement, as the patient was at high risk for bleeding. Likewise, due to the high risk of bleeding, the use of heparin as a supportive therapy was not advocated. The patient received a platelet transfusion, with an aim of a platelet count greater than 50 x 109/L. As well, the patient received fresh frozen plasma (FFP) and fibrinogen concentration, guided by the concentration in the patient’s plasma. Supportive treatment will continue to be maintained during remission induction of AML-M3, and the disappearance of the coagulopathy.
Discussion
The typical symptoms of DIC are generally those of the underlying process, which incites the condition. Underlying conditions may include, but are not limited to, severe infection with any microorganism, sepsis, trauma, solid tumors and malignancies, hepatic failure, and obstetric complications.4 Acute DIC may present as ecchymosis or petechiae. Additionally, there is usually a patient history of bleeding such as gastrointestinal or gingival bleeding.4 Post-operative DIC patients can also experience bleeding from various surgical sites, or within serous cavities.5 One should remain cognizant of signs and symptoms of deep vein thrombosis (DVT), as well as microvascular thrombosis. One hallmark of DIC is bleeding from at least three unrelated sites.4 Chronic DIC may present as thrombosis due to excess thrombin formation. Patients with comorbid liver disease may also present with jaundice, while patients with pulmonary involvement may present with a cough, dyspnea, and hemoptysis.4
Epidemiology
DIC occurs in all ages and races, and has not been shown to have a predisposition for a particular gender.4 DIC has been known to develop in about 1% of all hospitalized patients.4 DIC accounts for 9%-19% of intensive care unit (ICU) admissions.2 Although assigning values to DIC-related morbidity and mortality proves difficult, studies show it occurs in roughly 35% of septic patients.1 In a trauma setting, the presence of DIC may often double the mortality rate.4 DIC may be diagnosed in roughly 15% of patients with underlying acute leukemia.1 Previous studies report the occurrence of major bleeding to be 5%-12%, although patients with a platelet count of less than 50 x 109/L show a higher risk for bleeding when compared to patients with increased platelet counts.1 The occurrence of thrombosis in both small and midsize vessels is more commonly seen, which contributes to organ failure. Overall, the severity of DIC is directly related to increased mortality, thus emphasizing the importance of early recognition.
Related Disorders/Conditions
Several conditions are known to be complications of DIC. These conditions include, but are not limited to, kidney injury, altered mental status, respiratory dysfunction, hemothorax, gangrene, digit loss, and shock.4 In most cases, the release of tissue material into circulation in conjunction with endothelial disruption, leads to the activation of the coagulation cascade.
Pathophysiology
In DIC, the patient’s underlying condition stimulates powerful procoagulant activity that results in excess thrombin. The excess thrombin then overcomes the anticoagulant control mechanisms of protein C (PrC), antithrombin (AT), and tissue factor pathway inhibitor (TFPI), allowing for widespread thrombosis throughout the vasculature.2 The patient paradoxically battles between an excess thrombin state due to thrombin, embolism, and microvascular occlusion, and a hemorrhagic disorder secondary to depletion of platelets, consumption of coagulation factors, and accelerated formation of plasmin.2
To fully understand DIC, one must review normal hemostasis. Normal hemostasis generally takes place in response to an injury to the blood vessel wall. In normal hemostasis, a well-maintained process takes place in three stages: primary hemostasis, secondary hemostasis, and fibrinolysis. Primary hemostasis results in formation of the primary platelet plug via platelet adhesion, aggregation, and secretion.6 Secondary hemostasis is responsible for the formation of fibrin, and involves plasma proteins. In secondary hemostasis, fibrinogen is converted to fibrin, which forms a mesh that acts to reinforce the primary platelet plug, thus forming a hard, more stabilized clot at the site of injury, stopping any active bleeding.6 Finally, fibrinolysis takes place. Fibrinolysis is the process of the removal of fibrin through proteolysis, so that normal vessel structure and function can be restored.6 As part of normal healing, in response to an injury, clotting should only occur as a localized phenomenon, as there are many checks and balances to prevent extension of the hemostatic plug mechanism to the entire intravascular system.2 In DIC, this process of hemostasis runs out of control.
Laboratory Tests and Applications
Since DIC is secondary to an underlying disease, its diagnosis can be challenging, as there is no single laboratory test with sufficient accuracy to allow for a diagnosis. Imaging studies are generally only useful in detecting the underlying condition inciting DIC.4 DIC may present with a wide range of abnormalities in laboratory values. Laboratory findings of thrombocytopenia, prolonged coagulation times, decreased fibrinogen, elevated fibrin degradation products (FDPs), and elevated D-dimer levels are indicative of DIC.4 A complete blood count (CBC) and peripheral blood smear may show moderate-to-severe thrombocytopenia, as well as the presence of schistocytes secondary to evidence of microangiopathic pathology.4 Additionally, scoring systems have been developed to aid in diagnosis of DIC.4 The International Society on Thrombosis and Hemostasis (ISTH) developed a scoring system that makes use of laboratory tests, and a DIC score calculator. However, it is important to note, the presence of an underlying condition frequently associated with DIC is essential for the use of this diagnostic algorithm.4 Studies have reported the sensitivity of the DIC as 93%, and the specificity as 98%.4 As well, this scoring system has shown to be a strong predictor for mortality in sepsis. Similar scoring systems have been developed in other countries as well. Other conditions that may be ruled out by these tests include thrombotic thrombocytopenic purpura (TTP), hemolytic uremic syndrome (HUS), immune thrombocytopenia (ITP), chronic liver disease, Heparin-induced thrombocytopenia (HIT), and Coumadin/Vitamin K deficiency.2
Correlation of Laboratory Tests
Thrombocytopenia is the most common laboratory finding in DIC, reported in 93% of cases.5 In many cases, the thrombocytopenia may not yet be severe; therefore, it is crucial to recognize a downward trend in platelet count despite a recent count in a normal range.5 Acute DIC typically presents with a rapid consumption of platelets and clotting factors, so that at the time of diagnosis, the platelet count may be less than 50 x 109/L, the PT and aPTT values are markedly prolonged.2 FDPs appear elevated, as they are released into circulation as the body attempts to break down clots. Fibrinogen levels may be decreased, as it is used up. Likewise, plasminogen and AT show a decrease.2 D-dimers appear increased as a result of fibrinolysis and excess plasmin.2 Red blood cells (RBCs) that are pushed through occluded vessels in the microvasculature become fragmented and will often appear on a peripheral blood smear as schistocytes.
Impact of Pathophysiology on Laboratory Results
DIC can present with a variable range of abnormal laboratory values. Additionally, the values may only represent a momentary glimpse into a rapidly changing systemic process and often requires frequent repeated testing.4 As well, patients with chronic DIC may present with only minimal abnormalities in laboratory tests. There is no single test that is a specific indicator for DIC. Rather, the use of combined tests such as platelet count, PT, aPTT, fibrinogen, and D-dimer, in conjunction with an underlying disorder, help guide management and treatment.
A diagnosis of DIC is reached only through a combination of the clinical impression in conjunction with laboratory abnormalities.5 The patient history plays a significant role in that DIC is always secondary to an underlying condition, therefore the clinical presentation of DIC will be the features of the primary disease.
Treatment
The treatment of DIC typically involves focusing on the underlying disorder, as manifestations of DIC will subside once the underlying condition is addressed. Management of DIC itself follows four basic features: monitoring vital signs, assessing and documenting the extent of thrombosis and hemorrhage, correcting hypovolemia, and administering basic hemostatic procedures when indicated.4 Therapeutic interventions are mostly supportive in nature, and may include transfusion of blood components including platelets and clotting factors. However, blood components should be reserved for those patients who have hemorrhage, require a surgical procedure, or pose a high risk for bleeding complications.2 The use of heparin is usually reserved for cases of chronic DIC, and should be provided to patients demonstrating widespread fibrin deposition without evidence of substantial hemorrhage.4 Generally, the use of antifibrinolytic drugs should be avoided, as they have been known to cause thrombotic complications, although these agents have proven useful in cases associated with AML-M3 and other types of cancer.4 In these cases, Heparin should always be administered with antifibrinolytic drugs so as to arrest any prothrombotic effects.4 In patients with trauma and massive blood loss, antifibrinolytics such as tranexamic acid has shown to be effective in reducing blood loss and improving survival.4 Only minimal cases have been reported to benefit from administration of activated PrC.
Patient Response to Treatment
Treatment decisions for this patient are based on clinical and laboratory evaluation of hemostasis. The patient is to avoid all invasive procedures such as IV-line placement and biopsies. Heparin, in this case, is contraindicated secondary to bleeding risk. The patient received FFP, platelets, and fibrinogen concentrate. The patient currently remains hemodynamically stable, however, if evidence of significant bleeding and hyperfibrinolysis remains, tranexamic acid may be administered in conjunction with heparin. Clinical and laboratory parameters will continue to be monitored every eight hours.
Prognosis
The prognosis of patients with DIC depends on the severity of the coagulopathy, as well as the underlying condition that incited DIC. Generally, if the underlying condition is self-limiting or can be appropriately managed, DIC will disappear, and coagulation status will return to normal.4 Since DIC is a complex process, it is crucial to monitor the patient for clinical improvement or worsening, as well as to identify the early development of possible complications.
Treating the underlying AML-M3 is critical for the prognosis and management of this patient. The patient is encouraged to regularly consult with a hematologist for assistance with management of DIC, as chronic DIC may be treated on an outpatient basis after stabilization.4 Chronic DIC in patients with cancer may be managed with low molecular weight heparin or subcutaneous heparin. As well, other subspecialty consultations are necessary as indicated by the patient’s primary diagnosis.
Conclusion
DIC is not a specific illness, rather, it is a complication or an effect secondary to the progression of other underlying illnesses. It is associated with numerous clinical conditions, which involve excessive activation of the coagulation system.
Although interventions are supportive, they have been shown to be only partly effective. Since the clinical course of DIC ranges from asymptomatic to life-threatening, understanding its pathogenesis is vital for early recognition. Despite diagnostic criteria to aid physicians in the management of patients with DIC, newer laboratory markers and treatment modalities are warranted to improve patient outcome.
References
1 Levi M, Scully M. How I treat disseminated intravascular coagulation. Blood. 2018 Feb 22;131(8):845-54. doi: 10.1182/blood-2017-10-804096
2 Boral BM, Williams DJ, Boral LI. Disseminated intravascular coagulation. Am J of Clin Pathol. 2016 Dec;146(6):670-68. doi: 10.93/AJCP/AQW195
3 Medline Plus [Internet]. Bethesda (MD): National Library of Medicine (US); updated [date]. Disseminated intravascular coagulation; [updated 2022 Mar 21; reviewed 2019 Sep 24; cited 2022 Mar 30]; [about 2 p.] Available from: https://medlineplus.gov/ency/article/000573.htm
5 Thachill J. Disseminated intravascular coagulation: A practical approach. Anesthesiology. 2016 Jul;125(1): 230-6. doi: 10.1097/ALN.0000000000001123
6 Rodak BF, Fritsma MG, Fritsma GA. Normal hemostasis and coagulation. In: Hematology: Clinical principles and applications. St. Louis, MO: Elsevier Saunders; 2012. p. 626–44
7 Hussain SA, Zafar A, Faisal H, Vasylyeva O, Imran F. Adenovirus-associated disseminated intravascular coagulation. Cureus. 2021 Mar 30;13(3)e14194. doi: 10.7759/cureus.14194 8 Onishi T, Ishihara T, Nogami K. Coagulation and fibrinolysis balance in disseminated intravascular coagulation. Pediatr Int. 2021 Nov;63(11):1311-18. doi: 10.1111/ped.14684
-Kaysi Bujniewicz, MLS(ASCP)CM graduated Magna Cum Laude from University of North Dakota School of Medicine with a Bachelor of Science in Medical Laboratory Science. She has worked in clinical laboratories for over eight years as a certified and licensed Medical Laboratory Technician and a Medical Laboratory Scientist. Although her true callings are in Immunohematology and Clinical Microbiology, she currently works as a Generalist.
One of my favorite scary psychological thrillers is the movie, The Silence of the Lambs. The title was derived from a story told by the main character about her nightmares of screaming farm animals. A minor theme in the movie revolves around how much oversight the main character’s boss gives as she works on her first major crime case. The correct amount of oversight is key for any leader, especially inthe laboratory setting. Unfortunately, some leaders are all too silent and all too absent when it comes to safety oversight in their department.
In a laboratory, leadership is about more than just ensuring the smooth operation of daily tasks. It’s about creating a culture that prioritizes safety, fosters communication, and encourages accountability. When leadership is absent, whether physically or mentally, the ripple effects on safety can be devastating. Absentee leadership in laboratories is not a rare occurrence. With the demands placed on managers and supervisors, it’s easy to see how they might become disconnected from their teams. But when leaders disengage, lab safety is often the first thing to suffer.
Absentee leadership is often subtle, and it can be hard to recognize. It isn’t just about leaders being physically absent from the lab; it’s about them being emotionally and mentally checked out. These leaders are often unavailable for questions, slow to address concerns, or overly focused on administrative tasks that pull them away from the people they lead. This disconnection can lead to significant safety concerns.
Absentee leaders tend to neglect their role in enforcing routine safety protocols, assuming that staff will handle safety on their own. Without regular check-ins, inspections, or encouragement, safety lapses can become more frequent. Missing PPE, outdated safety procedures, and failure to address faulty equipment are all symptoms of a leadership vacuum. Over time, the lack of oversight can lead to more serious safety incidents, putting everyone at risk.
An absentee leader might miss the opportunity to reinforce the importance of safety or even dismiss safety concerns as unimportant compared to operational goals. This can create a culture where staff feel undervalued or believe that cutting corners is acceptable. Over time, low morale leads to disengaged employees, which can result in increased accidents, injuries, and near misses. After all, if leadership isn’t concerned with safety, why should the staff be?
Open and clear communication is essential for any safe work environment. However, absentee leaders tend to be unavailable when their team needs them. Safety concerns often go unaddressed because staff are unsure how or when to bring up issues. The lack of feedback and follow-up can lead to confusion about protocols, increasing the chances of mistakes. Without a strong communication chain, potential safety hazards are less likely to be reported and resolved quickly.
Ongoing training is vital in any laboratory to ensure that safety protocols are up-to-date and properly followed. When leaders are disconnected from their teams, they may fail to provide adequate training or overlook critical competency assessments. This can lead to staff being unaware of new safety procedures or forgetting essential protocols. Inconsistent training undermines the very foundation of a safe work environment, leaving gaps that could lead to accidents.
Recognizing absentee leadership is an important first step, but what’s most important is taking action to correct it. Being a leader in the lab means more than just sitting behind a desk in the office. Get out on the floor, engage with your team, and participate in day-to-day operations. This doesn’t just make you more visible; it makes you more approachable. Staff are more likely to bring up safety concerns if they see you regularly and know that you are open to hearing them. Make safety a regular topic of discussion in meetings, and let your team know that you are available for any questions or concerns they may have.
As a leader, you set the tone for the rest of the team. If you prioritize safety, team members will as well. Wear your PPE, follow safety protocols, and participate in safety drills. Show your team that safety is a personal responsibility for everyone, including you. By modeling the behaviors you want to see in your staff, you’re sending a clear message: safety isn’t just something we talk about—it’s something we do.
To keep safety at the forefront, establish regular safety check-ins with the staff. This could involve weekly safety huddles, monthly inspections, or one-on-one discussions about safety with each team member. Regular check-ins show that you are not only present but also actively engaged in maintaining a safe work environment. During these check-ins, ask specific questions about safety concerns and listen to what your staff have to say. This will help you stay informed about potential risks and demonstrate your commitment to safety.
An open-door policy is critical in preventing absentee leadership behaviors. However, it must be backed by action. Ensure that your team feels comfortable speaking up about safety issues without fear of retaliation. Make it clear that safety concerns will be addressed promptly and taken seriously. Additionally, follow up on reported issues and provide feedback on how they were resolved. This reinforces trust and lets your team know that their voices are heard.
Safety training shouldn’t be a one-time event. Ongoing education and competency assessments are crucial to maintaining a culture of safety. Review your team’s training records regularly and ensure that everyone is up-to-date on the latest protocols and procedures. Schedule routine refreshers on critical safety topics and offer training on new equipment or techniques as needed. By keeping safety training top-of-mind, you can help prevent complacency and ensure that everyone is equipped to work safely.
Lastly, leadership requires setting clear safety expectations and holding your team accountable for meeting them. Develop clear safety goals, such as maintaining a zero-incident work environment or completing a certain number of safety audits each month. Communicate these expectations clearly and provide regular feedback on how the team is performing. When safety expectations are consistently reinforced, they become ingrained in the lab’s culture.
Absentee leadership may not always be intentional, but its effects on laboratory safety are undeniable. When lab leaders are “silent,” they disengage, safety protocols slip, morale drops, and communication falters, leading to a more hazardous work environment. However, by taking proactive steps to become more vocal, engaged, present, and communicative, leaders can create a lab environment where safety is prioritized and consistently maintained. Leaders manage operations in the lab, but by not staying silent, they can creating a culture where safety nightmares become a thing of the past.
–Dan Scungio, MT(ASCP), SLS, CQA (ASQ) has over 25 years experience as a certified medical technologist. Today he is the Laboratory Safety Officer for Sentara Healthcare, a system of seven hospitals and over 20 laboratories and draw sites in the Tidewater area of Virginia. He is also known as Dan the Lab Safety Man, a lab safety consultant, educator, and trainer.
Language can have an indirect but profound impact on perception. The words we use to describe events can change how the act is interpreted, either consciously or subconsciously.
With that in mind, the way professionals and media speak about domestic violence (DV) often leaves much to be desired. Predictably, assaults and murders are described as “incidents” or “episodes”. This gentle substitution is a way to sanitize the physical and emotional trauma for a general audience. It is so commonly used that many professionals implicitly recognize ‘incident’ and ‘situation’ as a code for ‘abuse’. Yet these seemingly innocuous terms of convenience diminish the significance of the event. An “incident” is something small; a fender bender, or two people arguing in the security line at an airport. Tensions are raised but nobody is hurt, and things are soon back to normal. To label something an incident suggests it is of minimal importance to the general population. But when speaking about DV, this isn’t true. Unfortunately, the scope of the problem is incredibly wide. One study from the U.S. indicated 35.6% of women and 28.5% of men will be victims of DV in their lifetime (1). So even if you have not personally experienced it, you absolutely know someone who has (whether you know it or not). Every time someone is injured or murdered in a domestic violence “incident”, the entire community bears the loss – whether it is the loss of a friend, a parent, a co-worker, or all three.
In one relatively recent example, the District Attorney of Norfolk, Massachusetts described a double murder followed by suicide as a ‘domestic violence situation’ and a ‘domestic incident’. He stated it was ‘confined’ in the typically ‘nice neighborhood…safe community’. This ignores the obvious contradiction – safe community for most might be a better statement. But perhaps the most glaring mischaracterization is calling a crime of this nature ‘confined’. The intent is easy to interpret – there is no immediate threat to the safety of other residents – and that is an important piece of information. But to treat an act of violence so dismissively in a press conference is dishonest and diminishes the repercussions for the family, friends, and community of the victims. The effects reverberate through surviving family and friends in the community. There is nothing ‘confined’ about the effect of homicidal violence on a community. To speak about it that way treats the victims as though they were property of the perpetrator.
Relegating homicide to the category of ‘incident’ also dampens the emotional response to the loss. To place this in perspective, we would never describe the natural death of an elderly individual as a ‘cardiac incident’, or a fatal car crash as a ‘motor vehicle incident’. By allowing this compartmentalization, there is less public urgency to investigate the root causes and implement prevention efforts.
There are multiple tiers of responsibility. Media cannot do a better job reporting on these crimes if the professionals who autopsy, prosecute, and investigate cannot improve the language used. As one of the professionals who has used the term ‘incident’, I’m also trying to do better.
If you or someone you know is affected by domestic violence, please contact the National Domestic Violence Hotline at www.thehotline.org, or 1-800-799-SAFE.
References:
Black, M.C., Basile, K.C., Breiding, M.J., Smith, S.G., Walters, M.L., Merrick, M.T., Chen, J., & Stevens, M.R. (2011). The National Intimate Partner and Sexual Violence Survey (NISVS): 2010 Summary Report. Atlanta, GA: National Center for Injury Prevention and Control, Centers for Disease Control and Prevention.
-Alison Krywanczyk, MD, FASCP, is currently a Deputy Medical Examiner at the Cuyahoga County Medical Examiner’s Office.
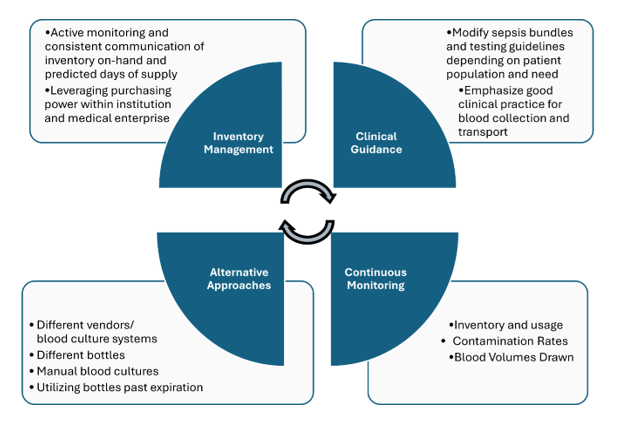
In June 2024, BD (Becton Dickinson) notified their customers that there will be a global shortage of BACTEC blood culture bottles which is anticipated to last several months. Blood culture bottles are critical for diagnosing infections in patients, and their scarcity could lead to delays in diagnosis and challenges in managing patient care, especially for those with serious systemic infections. During supply shortages, it is paramount that management relies heavily on careful inventory oversight, and prudent use and stewardship of available reagents. Here we will summarize key measures recommended to ensure continuity of care. However, it must be emphasized that there is no ‘one size fits all’ approach and each institution need to consider what works best for their patients and workflow. It is a team effort-laboratorians must work collaboratively with infectious disease providers, hospital leadership, supply management teams and other stakeholders to implement and manage efforts without duplication of efforts.
Figure 1. Summary of key measures used in dealing with blood culture bottle supply shortage
Inventory Management:
There needs to be continuous monitoring and communication among all stakeholders regarding inventory and usage. It is imperative that hospital leadership are aware of the current inventory on-hand and the predicted days of supply.
If the hospital is within a medical center, consider centralizing supplies and reallocating to those in dire need. If your institution is part of a larger system or corporation, consider leveraging system’s purchasing power vs. a single institution purchasing power to ensure availability of the blood culture bottles.
Clinical guidance:
In many institutions, laboratory and infectious disease teams, in collaboration with the clinical partners, developed sepsis bundles, in which blood cultures and lactic acid testing are the corner stones of the bundle. Most of the time, such bundles are built around the CMS guidelines, as part of the overarching Medicare’s Hospital Value-Based Purchasing Program (VBP). Such bundles are great tools to promote the best standard of care practices, as well as serving as a standard communication tool. Most organizations institute two separate types of sepsis bundles: the severe sepsis bundle and the sepsis shock bundle. With time, we need to consistently leverage quality and patient safety approaches, as well as regulatory standards to ensure the most optimal test utilization overall, especially during the times of shortages. Implementation of organization testing guidelines in the bundles promotes standardization and compliance.
Before ordering any tests for infectious diseases, it is crucial to carefully evaluate the pre-test probability of bacteremia and the likelihood of identifying a clinically significant organism. Repeating blood cultures within 48 hours of the initial set typically yields low results. Blood cultures should be collected before initiation of antimicrobial therapy. Indications for blood culture draws may vary depending on the patient population (e.g. adults versus pediatrics, immunocompetent versus neutropenic, inpatients versus from the emergency department) [1, 2]. For certain patient populations, consider performing only two blood cultures (1 set) vs. widely accepted and recommended four blood cultures (2 sets). While this is beyond the scope of this blog post, please refer to the sources below for specific clinical guidance.
Due to the typically low levels of bacteria present in the bloodstream, collecting a larger volume of blood increases the chances of obtaining a positive culture. To maximize the likelihood of detecting bacteria, blood culture bottles should be filled with the optimal volume recommended by the manufacturer, typically 8-10 mLs up to the fill line, and be sent to the microbiology laboratory in the timely manner. Reducing contamination in blood cultures decreases the need for additional resources, such as repeat blood cultures, unnecessary antibiotic treatments, and further diagnostic tests. Contamination can be minimized by strictly adhering to antiseptic procedures at collection sites, ensuring proper training for collectors. It is also crucial to remind healthcare providers and clinical staff to follow institutional guidelines for proper specimen labeling, maintaining specimen stability, and adhering to transport instructions to reduce the risk of sample rejection and prevent unnecessary waste of supplies [3].
Thinking outside the box:
Despite the shortage, patient care continues, and laboratories may need to think of alternative approaches. As of writing this post, other blood culture systems and their respective bottles are not affected. That said, implementing new instruments and switching to an alternative supply chain from a different manufacturer may be an option but often can take up to several months due to contractual requirements. Additionally, massive re-education and training (laboratory staff, phlebotomy, and nursing) will need to take place as different systems have unique collection procedures. Some institutions may opt to send out their blood cultures to reference laboratories that use an alternative system, although turnaround time may be an issue. Manual blood cultures, although laborious, could be set up and be done in-house. While it is not recommended to use expired blood culture bottles, recent published studies have shown that expired bottles up to 3 months are just as effective [4, 5]; institutions should maintain expired bottles if regulatory bodies allow for their utility at an later date as BD have extended the expiration date for certain lots already (see BD manufacturer webpage below). BD has recently announced the re-introduction of anaerobic media in glass bottles. Laboratories should prepare staff and develop plans for validation work to ensure that these bottles can be rapidly implemented for usage.
Continuous monitoring:
Aside from monitoring inventory and bottle usage, it is important for laboratories to regularly monitor contamination rates and implement a quality system that tracks blood culture volumes. To allow for targeted and more efficient education, monitoring trends stratified by different departments can help offer specific feedback to the clinical partners collecting the samples.
As we implement additional testing methodologies, remember to continuously revise your Clinical Practice Guidelines (CPGs), order sets, test utilization, etc. as part of such implementations.
A lesson learned from the COVID-19 pandemic is the need to be agile and quickly adapt to the changes in supply chain. For many laboratories, this was an effective strategy to ensure testing availability for all patient populations. Although this blood culture bottle shortage is a crisis many are facing, implementing diagnostic stewardship for blood culture collection may have overall benefits such as decreasing the amount of unnecessary testing, treatment of clinically insignificant infections, unnecessary blood draws, and potential central line-associated bloodstream infections.
Here are external resources/references available to help laboratories and healthcare providers:
Manufacturer’s web page: https://bdbactec-update.com/
American Society for Microbiology (ASM) guideline, endorsed by the Society for Healthcare Epidemiology of America (SHEA): https://asm.org/guideline/blood-culture-shortages-management-diagnostic-stew
Informational web page from the Infectious Disease Society of America (IDSA): https://www.idsociety.org/clinical-practice/blood-culture-bottle-shortage/
References
1. Fabre, V., K.C. Carroll, and S.E. Cosgrove, Blood Culture Utilization in the Hospital Setting: a Call for Diagnostic Stewardship. J Clin Microbiol, 2022. 60(3): p. e0100521.
2. Fabre, V., et al., Principles of diagnostic stewardship: A practical guide from the Society for Healthcare Epidemiology of America Diagnostic Stewardship Task Force. Infect Control Hosp Epidemiol, 2023. 44(2): p. 178-185.
3. Clinical and Laboratory Standards Institute, Principles and Procedures for Blood Cultures, CLSI guideline M47. 2nd Edition. 2022.
4. Hardy, L., et al., Blood culture bottles remain efficient months after their expiration date: implications for low- and middle-income countries. Clin Microbiol Infect, 2024. 30(10): p. 1327-1328.
5. Klontz, E.H., et al., Evaluation of expired BD BACTEC blood culture vials. J Clin Microbiol, 2024: p. e0108224.
-Rebecca Yee, PhD, D(ABMM), M(ASCP)CM is the Chief of Microbiology, Director of Clinical Microbiology and Molecular Microbiology Laboratory at the George Washington University Hospital. Her interests include bacteriology, antimicrobial resistance, and development of infectious disease diagnostics.
-Olga Kochar, MS, CSSGB, is the Divisional Director of Laboratory and Transfusion Services with the GW Hospital, as well as faculty with the GWU School of Medicine and Health Sciences. With over 29 years in the healthcare industry, Olga Kochar is passionate about quality and patient safety, performance improvement and teaching.
We do it several times a day, every day we work in the laboratory. We disinfect, but how much do we really think about what we are doing? Some may think we disinfect our benches at the end of the shift because it is expected, and that we only perform this task because it is listed on our maintenance log. Others disinfect at the beginning and at the end of their shift because of how they were trained. Since the pandemic, I bet many lab techs, nurses, and hospital staff disinfect religiously out of fear of catching COVID. The real reason we disinfect is to keep ourselves, our coworkers, and the people we care about safe.
As laboratorians, we have practices to preemptively and proactively ensure that the samples and patients we work with do not put us in harm’s way. We clean and disinfect anytime we have a spill of patient specimen. With that spill we know there is a potential for exposure, and we take steps to remove the threat. We also routinely disinfect our work areas throughout the day to eradicate any unseen pathogens. It is easy to visually spot a spill of whole blood, but a few drops of serum or plasma can easily go undetected.
To keep everyone safe, we need to fully comprehend the difference between “cleaning” and “disinfection.” Cleaning is the act of removing the physical component of a spill, sample, or mess. This could be dirt, dust, blood, body fluid, or even a piece of tissue. If there is a spill of blood on the floor, we first clean up the spill by applying an absorbent and sweeping up the material. We can see that the blood is removed from the floor, but what we cannot see poses the most danger. Simply cleaning up a spill does not kill any potential pathogens. We still have one more step, disinfection.
Disinfection is the process of killing the pathogens and it must take place after cleaning. Only after we disinfect is the area considered safe. It takes both the cleaning and disinfection steps to make sure the spill is properly handled. Spraying bleach directly on a blood spill and wiping with a paper towel may leave pathogens under the spill lurking on the surface of the floor or counter. For disinfection to be the most effective, it is essential to expose the potential pathogens to the disinfectant, and cleaning helps remove any barriers.
We have the why behind the process, so let’s discuss how we disinfect. Our best defense against most pathogens is bleach. Ah, the glorious smell of bleach; either you love it or hate it. Bleach is one of the few products that can effectively neutralize Clostridium difficile. That is why bleach is used for benchtop and post-spill disinfection. In the lab setting, we typically see two types of bleach products, a spray and wipes. Each has their own minimal contact time, so make sure to leave the product on the area for the allotted amount of time. Bleach wipes typically require a longer contact time than bleach spray. It is also important to make sure the lids on any disinfectant wipe container remain closed when not in use. If the lids are left open, the wipes can dry out, making them a less effective disinfectant.
Some labs may allow for a bleach alternative when employees with a bleach allergy are present in the lab. Individuals with a documented bleach allergy may be permitted to use alternatives such as a concentrated broad-spectrum quaternary agent as a means for disinfecting. Usually, approval must come from an organization’s employee health or physician to accommodate the request. As with bleach wipes, quaternary disinfectants may require a longer contact time than your usual bleach spray.
You may see products from other manufacturers that use alcohol with an ammonium chloride ingredient. Many labs use these types of disinfectants in areas to eliminate the residue seen when using a bleach product, such as cleaning stainless steel biological or chemical safety hoods, glass, or other office appliances. Often, alcohol or ammonium chloride disinfectants are used on telephone headsets, computer screens, select Point of Care instruments, and other items for which bleach is prohibited. In addition, some instrument manufacturers recommend only using a particular product to clean the surface of their equipment to ensure the cleaning does not damage the outer casing or internal components. Sometimes, though, staff will accidentally substitute these types of products in place of bleach when it is time to disinfect at the end of the shift or help with a spill. It may be convenient to just grab a bottle of an alcohol-based disinfectant and jump into action. However, it is more important not to take short cuts and locate a bottle of bleach spray and disinfect the proper way. Sometimes it may seem like a hassle, but disinfection is part of our laboratory culture, and we need to keep up with the practice. If we maintain our disinfection routines, there is no reason to doubt our safety inside and outside of the lab. You can take it a step further and always be on the lookout for expired products or open lids to ensure your products can do their jobs effectively. Understanding why we perform these tasks and the importance they have for our safety is something that should be shared with all employees. Together, with the right mindset, you can be confident that your lab is a safe, clean, and properly disinfected lab.
-Jason P. Nagy, PhD, MLS(ASCP)CM is a Lab Safety Coordinator for Sentara Healthcare, a hospital system with laboratories throughout Virginia and North Carolina. He is an experienced Technical Specialist with a background in biotechnology, molecular biology, clinical labs, and most recently, a focus in laboratory safety.
Inspections are a great learning opportunity. Our recent American Association of Blood and Biotherapies (AABB) introduced us to the risk analysis requirement in its 2024 checklist. Though the requirement is specifically stated in 3.7 Information Systems #13, the requirement can be applied to instruments as well as software. The update is significant as it emphasizes the need to proactively identify, assess, and mitigate risks associated with introducing or modifying software and instruments within the laboratory setting.
AABB-accredited laboratories will need to review their validation procedures to ensure they include a risk analysis with mitigation for identified risks. There are a few steps to conducting a risk analysis.
First, clearly define the changes being made. This could include new software installations, updates to existing software, introduction of new instruments, or modifications to current equipment. Understanding the full scope of changes is essential for a comprehensive risk analysis.
Secondly, if possible, form a multidisciplinary team that includes IT specialists, laboratory managers, quality assurance personnel, and end-users. In reality, for many small to medium laboratories, frequently one person fills more than one role. It’s conceivable for the laboratory manager to be the quality person and an end user since they often may have to fill in during staff shortages.
Next, identify the risks. Structured techniques like Failure Modes and Effects Analysis (FMEA) can be used to identify potential risks. Consider software-based risks such as data loss or corruption, system incompatibility, user interface issues, and cybersecurity vulnerabilities. Calibration errors, operational failures, compatibility with existing systems, and maintenance requirements are some instrument-based risks that should be considered. And, of course, human-based risks involving user errors, poor or insufficient training, and workflow disruptions.
The fourth step would be to assess each identified risk, assessing its potential impact on laboratory operations and patient safety and evaluating the likelihood of occurrence and the severity of consequences if the risk materializes. Use a risk matrix to categorize risks as low, medium, or high.
For each high or medium risk identified, develop mitigation strategies to reduce the likelihood of occurrence or minimize the impact. Some examples are increasing training programs, additional testing, or developing contingency plans for failures.
Ensure the entire risk analysis process is documented, including the identified risks, evaluation results, and mitigation strategies. Documentation is crucial for compliance with the AABB checklist and is a reference for future audits or inspections.
The risk analysis requirement in the AABB’s 2024 checklist underscores the importance of proactive risk management in clinical laboratories. Through the implementation of the outlined steps, laboratories can not only meet this requirement but also enhance their operational resilience and commitment to patient safety. Conducting a thorough risk analysis for software and instrument changes is an investment in the quality and reliability of laboratory services, ultimately contributing to better patient outcomes.
-Darryl Elzie is the Regulatory Affairs Manager Inova Blood Donor Services. He has been an ASCP Medical Laboratory Scientist for over 25 years, performing CAP inspections for two decades. He has held the roles of laboratory generalist, chemistry senior technologist, and quality consultant. He has a Master’s in Healthcare Administration from Ashford University, a Doctorate of Psychology from The University of the Rockies, and is a Certified Quality Auditor (ASQ). Inova Blood Donor Services is the largest hospital-based blood center in the nation. Dr. Elzie is also a Counselor and Life Coach at issueslifecoaching.com.
Back in the olden days – wait, can I use that line yet? I graduated from my cytology program ten years ago, which feels like two decades ago or just yesterday. Depends on the day. Anyway, there are little nuggets from my training that stick with me whenever I’m screening a case. This case revolves around the concept of a two-cell population in body fluids, namely pleural, peritoneal, and pericardial. We were taught that if you see a two-cell population, the case is malignant. I remember asking in school, “But what if the diagnosis is mesothelioma?” “Okay,” my professor said, “that applies to any malignancy except mesothelioma. The two-cell population indicates a population of benign mesothelial cells and a second population of malignant cells.” Noted! I live by this rule, and every now and then, I have a case of a peritoneal fluid where the slides are covered in “wall-to-wall” adenocarcinoma, and I can’t find a benign mesothelial cell for the life of me. Obviously, we let years of experience with morphology take the reins there, but there are still those cases where the native mesothelial cells are so reactive in appearance that we start hunting for a two-cell population. This case was a perfect blend of wall-to-wall tumor and more-than-reactive mesothelials.
When a patient is being worked up for the first time or the nth time, we recognize their names and recall their clinical history and previous morphologies. Quite often we have patients with genetic predispositions to cancer who present with one type of cancer and are treated accordingly, and during surveillance imaging, come back to us with a second primary cancer. In this case, we received a right-sided pleural fluid on a 73-year-old woman with a history of ER+/PR+/HER2- breast cancer. After a history of incomplete medical follow-up, the patient presented to a local emergency room with pleuritic pain, upon which a CT Scan identified a lung mass, breast mass, and multiple liver and upper thoracic bone lesions. The assumption, given the clinical history, was metastatic breast cancer, and the clinician submitted the pleural fluid for cytology to repeat ER/PR/HER2 testing to determine if there is a better treatment strategy than her current aromatase inhibitor, which appeared to be failing.
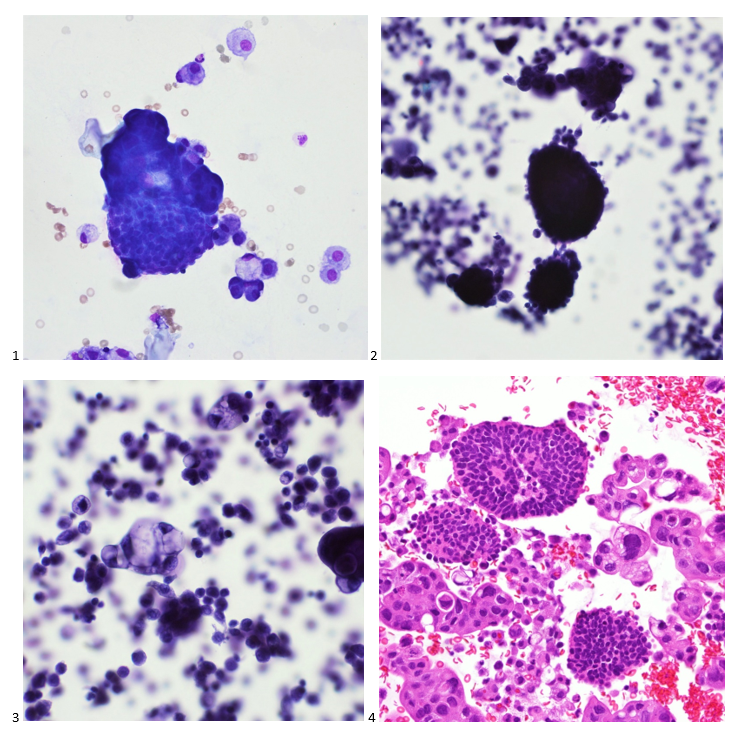
Upon screening the cytopreparations, we observed very obvious malignant cells (Image 1). It was evident that we had a two-cell population. One of the cell populations was characterized by cohesive clusters of relatively uniform tumor cells (Image 2 & 4) and the second cell population was less cohesive with larger pleomorphic cells (Image 3 & 4). Hmm, something doesn’t make sense here. We already have a group of tumor; the other cells are supposed to be benign mesothelials cells. Is there a mixed morphology going on? Like a combined small cell carcinoma and non-small cell carcinoma that we might see in lung cancer? It’s not small cell though. These two populations are clearly two adenocarcinomas. That still can’t be. There has to be a population of mesothelials. No, no… the benign mesothelials are scattered throughout the background. It looks like a double-malignant three-cell population!
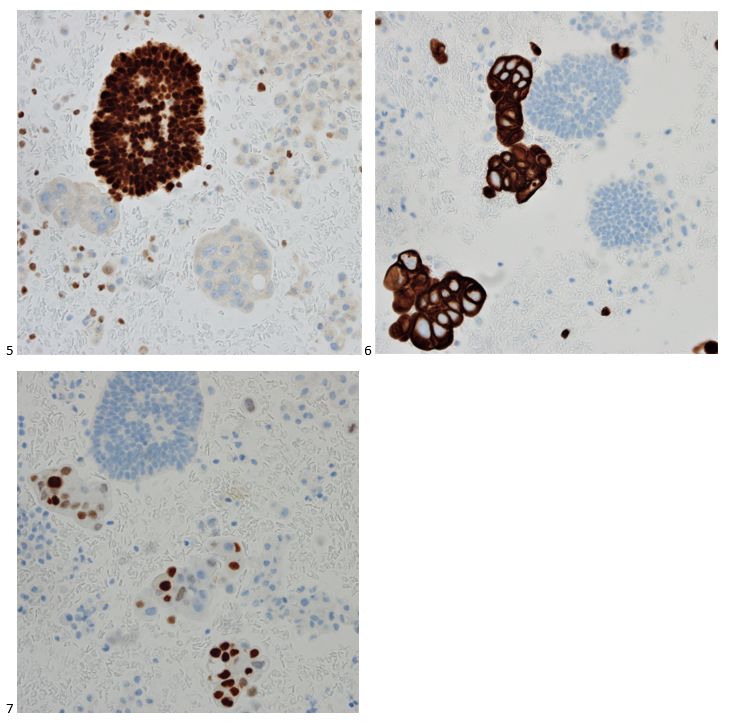
We submitted the case to the cytopathologist on service for the day. When they asked what we had, we replied, “All the adenocarcinoma,” and noted that we definitely need ER/PR/HER2 as clinically and now morphologically, it appears that the patient’s breast cancer is involving the pleural fluid. The cytopathologist agreed that there was something to the case. Metastatic breast cancer likes to appear as cannon balls in fluid (Image 2), the second cell population is too unlike the first. The cytopathologist performed immunocytochemical stains on paraffin sections of the cell block with adequate controls. The first type of cells shows positive staining for CK7, CK19, GATA3 (Image 5), and ER, and negative staining for CK20 (Image 6), CDX-2 (Image 7), BRST2, and PR. The second type of tumor cells show positive staining for CK7, CK20 (Image 6), CK19, and CDX-2 (Image 7), and negative staining for GATA3 (Image 5), ER, PR, and BRST2. The former group of cells has a morphology and immunoprofile consistent with breast origin, and the latter group of cells are morphologically and immunophenotypically suggestive of a pancreaticobiliary or gastrointestinal tract origin.
Final Diagnosis: Positive for malignancy. Adenocarcinoma, consistent with metastatic breast and metastatic pancreaticobiliary or gastrointestinal tract origin.
Interestingly, the patient had a paracentesis and liver biopsy the following week, and only the pancreaticobiliary or GI tract origin immunoprofile was positive in the peritoneal fluid and FNA. The patient’s breast cancer seemed to remain above the diaphragm.
-Taryn Waraksa-Deutsch, DHSc, SCT(ASCP)CM, CMIAC, LSSGB, is the Cytopathology Supervisor at Fox Chase Cancer Center, in Philadelphia, Pennsylvania. She earned her master’s degree from Thomas Jefferson University in 2014 and completed her Doctorate of Health Science from Bay Path University in 2023. Her research interests include change management and continuous improvement methodologies in laboratory medicine. She is an ASCP board-certified Specialist in Cytology with an additional certification by the International Academy of Cytology (IAC). She is also a 2020 ASCP 40 Under Forty Honoree. Outside of her work, Taryn is a certified Divemaster. Scuba diving in freshwater caverns is her favorite way to rest her eyes from the microscope.