Clinical History
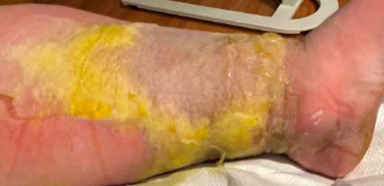
A 35 year old male with chronic bilateral lower extremity lymphedema due to obesity presented with a one-week history of subjective fevers and malaise with associated left lower extremity pain, swelling and erythema. The left leg was markedly edematous with erythema present above the knee down. The leg was tender to palpation, and multiple ruptured bullae and areas of severe desquamation with excessive serous drainage were observed. Importantly, no areas of purulence were noted (Image 2). A clinical diagnosis of severe non-purulent cellulitis was made, and the patient was admitted for parenteral antibiotic therapy of vancomycin and piperacillin-tazobactam. Necrotizing fasciitis was ruled out based on imaging, and significant clinical improvement was seen after 5 days of intravenous antibiotics. The patient was transitioned to oral therapy with amoxicillin-clavulanic acid and doxycycline for a total of 14 days of antibiotics.
Laboratory Workup
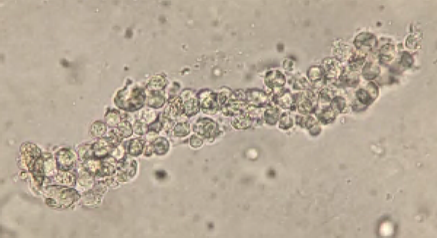
During the admission, urinalysis revealed turbid urine with elevated protein (30 mg/dL), and 2+ blood with 5 RBC/HPF on microscopic examination. Given the presence of protein with microscopic hematuria, causes of glomerulonephritis were investigated. Workup revealed a markedly elevated anti-streptolysin O (ASO) titer of 5310 (0-330) and a total complement (CH50) level of 14, which was low given his age. Urine sediment examination revealed red blood cell casts (Image 3). These clinical and laboratory findings were consistent with post-streptococcal glomerulonephritis (PSGN) due to Streptococcus pyogenes skin and soft tissue infection.

Image 1. Colony appearance and biochemical testing of S. pyogenes. A) Typical gram positive cocci in chains characteristic of streptococci. B) Growth on Sheep’s Blood Agar of small, translucent colonies with a wide zone of beta-hemolysis indicative of S. pyogenes. C) Catalase-negative S. pyogenes (left) compared to catalase-positive S. aureus (right). D) PYR-positive S. pyogenes (left) compared to PYR-negative S. aureus (right).
Discussion
Streptococcus pyogenes are gram positive bacteria that appear in pairs and/or chains by microscopy (Image 1A). In culture, these organisms produce relatively small colonies which elaborate a large zone of beta hemolysis on blood agar plates; colonies are translucent with smooth edges (Image 1B). The beta-hemolytic activity of S. pyogenes is due to the activity of two hemolysins: Streptolysin-S (oxygen-stabile) and Streptolysin-O (oxygen-labile). S. pyogenes is the primary organism which expresses the Lancefield Group A carbohydrate antigen. Less frequently encountered strains of S. anginosus and S. dysgalactiae subsp. equisimilis may also express this antigen, so biochemical identification of S. pyogenes may be helpful for a definitive diagnosis. MALDI-TOF MS may also fail to discriminate between S. pyogenes and closely related β-hemolytic streptococci (including S. dysgalactiae and S. canis), necessitating adjunctive biochemical testing. Like other streptococci, S. pyogenes is catalase negative (Image 1C). Unlike other beta-hemolytic streptococci, S. pyogenes expresses pyrrolidonyl arylamidase (PYR) making this test a rapid and useful adjunctive diagnostic tool (Figure 1D). Bacitracin susceptibility was used historically but has been largely replaced by PYR testing due to concerns over specificity and prolonged turnaround time.
Globally, S. pyogenes is responsible for a large percentage of infection-related morbidity and mortality. The organism colonizes the skin and the nasopharynx of humans, but most colonized individuals do not develop active disease. Colonization however can lead to infection or dissemination to susceptible individuals. S. pyogenes infections exhibit a diverse range of clinical manifestations which can include pharyngitis, impetigo, erysipelas, cellulitis, necrotizing fasciitis, pyomyositis, streptococcal toxic shock syndrome, and bacteremia. S. pyogenes remains susceptible to penicillin, making β-lactams first-line drugs of choice for management. Conversely, rising levels of macrolide, lincomycin, tetracycline, and fluoroquinolone resistance has been observed. Susceptibility testing may be warranted if these agents are to be used, most often in the cases of severe penicillin allergy.
S. pyogenes infection can be complicated by multiple post-infectious immune-mediated sequelae including PSGN and rheumatic fever. Post-Streptococcus glomerulonephritis (PSGN) has a global incidence of > 470,000 individuals per year and occurs due to the deposition of immune complexes in the glomeruli resulting from previous S. pyogenes pharyngitis or soft tissue infection (as seen in this case). Typical clinical presentation of PSGN includes hematuria, proteinuria, edema, hypertension, elevated serum creatinine levels, hypocomplementemia, and general malaise. The elevated ASO titer (5310) was diagnostic of an S. pyogenes acute infection as the cause of this patient’s cellulitis. The development of proteinuria and hematuria following infection further supports a clinical diagnosis of PSGN. Treatment of PSGN is largely supportive with the focus on management of the underlying infection. Most individuals with kidney failure from PSGN recover to baseline renal function; however, there may be a link between PSGN and the later development of chronic kidney disease/end-stage renal disease.
References
- De la Maza LM, Pezzlo MT, Bittencourt CE, Peterson EM. 2020. Color Atlas of Medical Bacteriology, 3rd edition. ASM Press. Pg. 11-23
- Madaio MP, Harrington JT. 2001. The diagnosis of glomerular diseases: acute glomerulonephritis and the nephrotic syndrome. Arch Intern Med. 161(1):Pg. 25-34. doi: 10.1001/archinte.161.1.25.
- Stevens DL, Bisno AL, Chambers HF, Dellinger EP, Goldstein EJC, Gorbach SL, Hirschmann JV, Kaplan SL, Montoya JG, Wade JC. 2014. Practice Guidelines for the Diagnosis and Management of Skin and Soft Tissue Infections: 2014 Update by the Infectious Diseases Society of America. Clin Infect Dis. 59(2): Pg. e10-e52, https://doi.org/10.1093/cid/ciu296.
- Walker MJ, Barnett TC, McArthur JD, Cole JN, Gillen CM, Henningham A, Sriprakash KS, Sanderson-Smith ML, Nizet V. 2014. Disease manifestations and pathogenic mechanisms of Group A Streptococcus. Clin Microbiol Rev. (2): Pg. 264-301. doi: 10.1128/CMR.00101-13.
- Wong CH, Khin LW, Heng KS, Tan KC, Low CO. 2014. The LRINEC (Laboratory Risk Indicator for Necrotizing Fasciitis) score: a tool for distinguishing necrotizing fasciitis from other soft tissue infections. Crit Care Med. 32(7): Pg. 1535-41. doi: 10.1097/01.ccm.0000129486.35458.7d.
-John Markantonis, DO is the former Medical Microbiology fellow at UT Southwestern and has recently completed his clinical pathology residency. He is also interested in Transfusion Medicine and parasitic diseases.

-Andrew Clark, PhD, D(ABMM) is an Assistant Professor at UT Southwestern Medical Center in the Department of Pathology, and Associate Director of the Clements University Hospital microbiology laboratory. He completed a CPEP-accredited postdoctoral fellowship in Medical and Public Health Microbiology at National Institutes of Health, and is interested in antimicrobial susceptibility and anaerobe pathophysiology.

-Clare McCormick-Baw, MD, PhD is an Assistant Professor of Clinical Microbiology at UT Southwestern in Dallas, Texas. She has a passion for teaching about laboratory medicine in general and the best uses of the microbiology lab in particular.