The FDA is now democratizing the testing of the novel coronavirus: SARS-CoV-2 (the virus which causes the COVID-19 disease syndrome—I will call it COVID-19 from here on as that is the colloquial name most people know) by allowing high complexity testing labs across the United States. This move will permit more labs to test for COVID-19. A previous post by contributor Constantine Kanakis describes the biology of the virus, so I will not repeat that material. Instead, I will focus on some considerations in validating a Lab Developed Test (LDT) COVID-19 molecular assay.
The president of AACC, Carmen Wiley, said there are 11,000 high complexity testing labs in the US, which could qualify for performing this testing. However, not all of these labs have molecular and virology expertise, so others have placed the number of labs with qualified staff and instrumentation at 400.
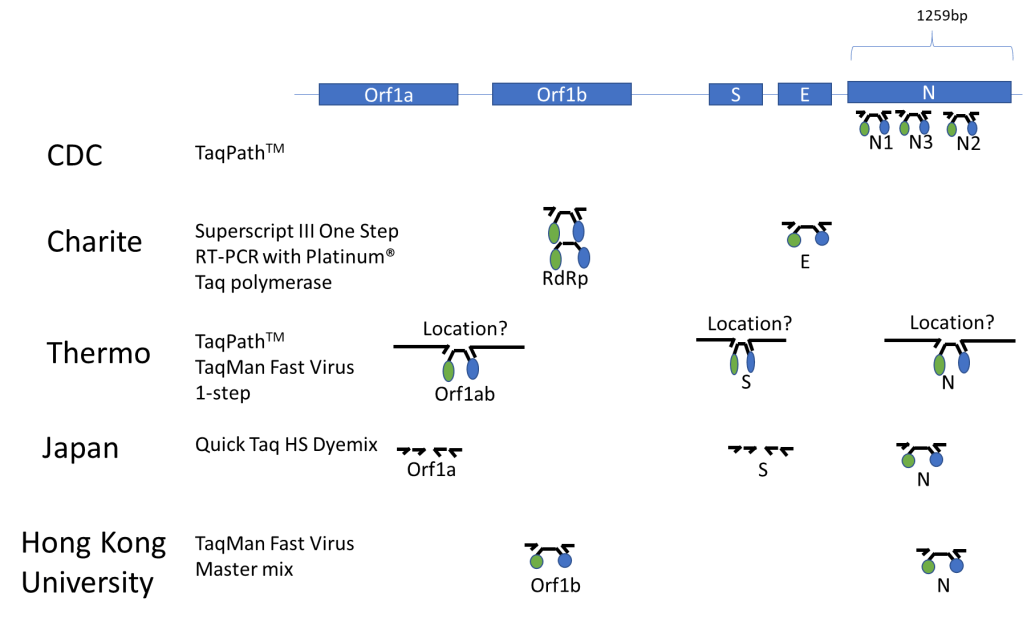
Published Assays and Targets: As an overview, the figure below (Figure 1) summarizes some published COVID-19 assays. As you can see, the major strategy involves using the TaqMan probe strategy where a short probe is degraded by Taq polymerase releasing a fluorescent molecule (green ball) from a quencher molecule (blue ball). The TaqMan approach allows for quick performance of the assay and easy interpretation. One lab from Japan is using nested PCR amplification and sequencing of the Orf1a and S genes as well.
In silico Cross-reactivity:
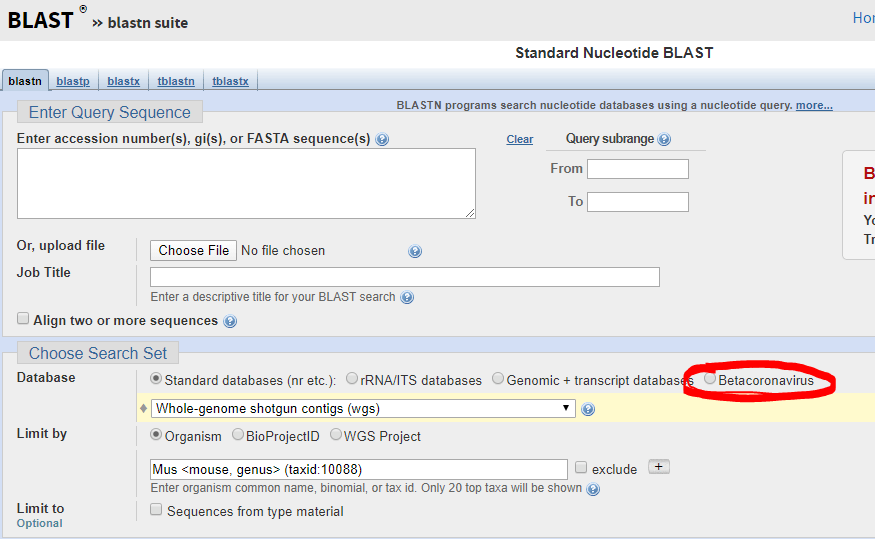
The FDA guidance allows cross-reactivity to be minimally assessed in silico by demonstrating “greater than 80% homology between primer/probes and any sequence present in the targeted microorganism.” The primer locations can be found in the publication of each protocol (except Thermo) and can be confirmed by checking the NCBI Blast site and they actually have a selection for beta-cornavirus (Figure 2) now that allows you to search for your primer’s reactivity across other related viruses- Very helpful!
Primer/Probe Design:
The N region is the most popular site to probe and is included in most kits once and the CDC kit three times. It was the reagent set for N3 in the CDC kit that was having difficulties, so you may decide to not include that component in your LDT. If you want to see how the different available primer sets align on the N gene sequence you can see below for the primers labeled based on their source. Many are overlapping, perhaps because many people thought the same site was a good target (Figure 3).

Commercially Available Assays:
An important part of validating your COVID-19 assay is to do so quickly. Thus commercially available kits would be helpful, however there are only two commercially available sources at this time: IDT and Thermo. IDT is producing a kit with the CDC design. Thermo produced their kit over the last few months and does not have any published validation information that I could find. Also Thermo when I checked just now for the catalog number, it says this product is unavailable… not sure what that means, but maybe you can try contacting them. Both IDT and Thermo list control plasmid reagents for their assays.
Controls for the Assay:
The wording of the FDA announcement was interesting in that it 1) did not require clinical samples, but allows “contrived clinical specimens.” “Contrived reactive specimens can be created by spiking RNA or inactivated virus into leftover clinical specimens.” A major difficulty is the access to actual COVID-19 RNA or inactivated virus. I noticed that the guidance didn’t say that the assay MUST use RNA. Thus most labs would have access to plasmid DNA, which could potentially be used.
Given the limited availability of RNA for validation use, a lab may consider performing much of the assay optimization with COVID-19 Plasmid DNA while waiting for access to RNA. I would like to be sure my assay could extract, amplify and detect RNA as part of the clinical validation.
Asuragen can produce Armored RNA, with synthetic RNA packaged inside of a viral capsid, which would be a useful control for extraction, amplification and detection. However, we heard this will not be available for another month.
Tom Stenzel (director of the Office of In Vitro Diagnostics and Radiological Health at the FDA’s Center for Devices and Radiological Health (CDRH)) said FDA, BARDA, and the CDC will prioritize and coordinate shipments of viral materials to labs when they are ready to validate tests according to a webinar with labs on Monday. Currently, the FDA is directing inquiries to BEI, which is reportedly prioritizing requests to send out samples in 12-72 hours.
Lastly, one could try to use in vitro synthesized RNA sequences surrounding your primer targets as a control for now and may have better luck in getting the product soon. This is the control that is being shipped with the CDC kits to public labs.
Limit of Detection is an unknown for what is likely to be clinically relevant as we don’t know what the levels look like in people with early vs. late vs. severe vs. mild disease. The FDA just says you should be able to detect 95% of samples (19 of 20) that are x1-x2 the limit of detection.
FDA Notification:
This is the final and important step. Once you go live, you must notify the FDA with an Emergency Use Assay (EUA) form within 15 days. Reviewing the form, there doesn’t appear to have complex explanations or overdue requirements for reporting, which wouldn’t be found in a standard lab validation document.
Final Thoughts/Future commercial solutions:
This information is the best of what I know right now based on current information- this is not a complete guide and the FDA guidance should be read closely for all compliance details. Information is changing quickly and is likely to change more if the number of COVID-19 cases in the United States increases. Cepheid, Luminex, and BioFire are reportedly working on assays that will be out in several months and would be easy to use for many labs that already have one or both of these systems-however it may require a full validation for an LDT, but I’m not sure as it is an EUA-further clarification on this point is needed. Although there are several commercial solutions available, we don’t know how demand could impact supply from each company. Fortunately, some large reference labs like LabCorp and Quest are looking to develop a COVID19 test. Good luck, stay safe, and feel free to contact me with any questions in the comments below so that everyone can benefit from the discussion!
References
In lieu of a list of references, I’ve included web links for the most current and direct sources of information.

-Jeff SoRelle, MD is a Chief Resident of Pathology at the University of Texas Southwestern Medical Center in Dallas, TX. His clinical research interests include understanding how the lab intersects with transgender healthcare and improving genetic variant interpretation.




