This is the first in series of “5 Things to Know” updates on various Transfusion Medicine topics, posted on Lablogatory and the Blood Bank Guy website. Today, we will cover a topic that is a mystery to many: Granulocyte transfusions!
1. We aren’t totally sure that granulocytes actually work! In the nearly half-century that granulocyte concentrates have been transfused, numerous retrospective articles, case reports, and small series about granulocytes have been published. If you ask Hematology-Oncology docs, you will likely find a general consensus that granulocytes can be effective (including a really cool, almost miraculous-sounding story!). Sadly, stories are not evidence. A study begun in 2008 called The “RING study” (Resolving Infection in people with Neutropenia with Granulocytes) was designed to gather evidence by randomizing recipients into either a group that received granulocytes or one that did not. Unfortunately, the study failed to gather enough participants before closing in 2013 to be truly meaningful (though the data that was gathered did not show a substantial beneficial effect, according to data presented at the 2014 American Society of Hematology Annual Meeting). 
So, does this mean that granulocyte transfusions are stupid? Not from my perspective! Honestly, if the standard for producing a product is proof positive that the product is effective, we blood bankers would have NOTHING on our shelves! Some of my colleagues disagree with me, but from my perspective, granulocytes have value in some limited settings (outlined below).
2. Modern granulocyte collections are a multiple day process. Historically in the United States, granulocytes were collected by apheresis either from “walk-in” unstimulated donors (more common) or from donors given a single dose of oral steroids to increase their white blood cell count. The unstimulated collections resulted in a product with a total granulocyte count of about 1.0 x 1010 (that number is the minimum requirement in at least 75% of collections, according to AABB Standards, and is a GREAT number to remember if you are studying for an exam!). However, if we have learned anything about granulocytes, we know that 1.0 x 1010 just isn’t enough! A “modern” U.S. granulocyte collection is from an apheresis donor stimulated the day before collection with an injection of Granulocyte Colony Stimulating Factor (“G-CSF”), in many cases supplemented by an oral dose of steroids (typically dexamethasone); this regimen typically results in a yield of 4.0 x 1010 granulocytes or more (please note that G-CSF is not FDA-approved for use in stimulating donors, so donors should have a formal informed consent prior to undergoing stimulation). As an aside, granulocytes may be produced in different ways outside of the US. The United Kingdom, in particular, does not permit G-CSF or steroid stimulation of granulocyte donors that are not family or friends of the patient. As a result, non-family member UK granulocyte products are produced from pooled buffy coats from ten random donors, with a yield of approximately 1.0 x 1010 granulocytes. These collections require lots of extra effort by numerous people (including our donors!), which leads us to number three…
3. Granulocytes should only be ordered in specific clinical settings Granulocyte collections are not easy, and they should not be ordered carelessly. While there is no universally agreed-upon set of conditions, granulocytes are most commonly utilized in one of the following clinical situations:
- Patients with hematologic malignancies and low neutrophil counts due to chemotherapy
- Stem cell transplant recipients during pancytopenic phase
- Neonates with sepsis
- Patients with chronic granulomatous disease
This is not to say that everyone in one of those situations will or should receive granulocytes. Instead, we are looking for some specific clinical and laboratory findings before agreeing to start the granulocyte collection process, especially in the first three groups of patients on the list above. Here’s what we like to see:
- Proven or highly probable bacterial or fungal infection (NOTE: Available data suggests granulocytes work better with bacterial infections)
- No response to appropriate antimicrobial therapy
- Absolute neutropenia (<500 granulocytes/microliter)
- A reasonable expectation that the patient will begin producing granulocytes soon
Traditionally, blood bankers have strongly resisted “prophylactic” granulocyte transfusions (for immunosuppressed patients without current evidence of infection but at high risk of acquiring one) or use in fever of unknown origin. There has been some debate about this recently, but most still believe (as do I) that granulocytes should only be given for patients who have an infection.
4. Granulocytes look funny, and have unusual storage, matching, and modification requirements. If you have never seen a granulocyte product, you might guess that granulocyte and platelet concentrates look alike, since both cells reside in the “buffy coat” and both are collected in the US primarily by apheresis technology. Your guess would be wrong! Apheresis granulocyte units typically contain between 30 and 50 mL of RBCs, because it is essentially impossible to get a good granulocyte yield without harvesting some RBCs as well (they are immediately adjacent to each other by density separation in the apheresis machine). 30-50 mL sounds like a small amount, but that quantity makes the granulocyte product look almost as “red” as a unit of RBCs! (see images below)


Notice how the same 30 mL that makes the bottom of the bag look red during collection (on left) makes the whole bag look REALLY red after mixing!
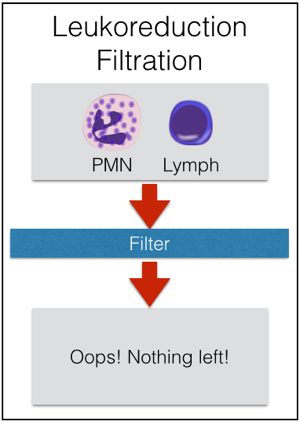
Granulocytes have pretty much the shortest shelf life of any product that we collect directly from donors. They should be transfused “as soon as possible” after collection, but definitely within 24 hours of collection. They are stored at room temperature, like platelets, but they should not be continuously agitated, unlike platelets. This out-of-the-ordinary storage is really another reason that granulocytes are only collected on an “as-needed” basis. Granulocytes are also unusual in that they are almost always issued prior to the availability of the infectious disease screening results! For that reason, essentially all unrelated apheresis granulocyte donors will be recent apheresis platelet donors who have had negative results on a donation within the previous 30 days. We still perform the testing, of course, but the results just aren’t available before the product has to be transfused. Because there are so many RBCs in each granulocyte product, the donor must be ABO compatible with the recipient, and the unit must be crossmatch-compatible with the recipient. By AABB Standards, if more than 2 mL of RBCs are present in any product, those RBCs must be compatible with the recipient’s plasma antibodies. In addition, if the recipient has an unexpected RBC antibody (like anti-D, anti-K, or any others), the granulocyte unit must come from someone who is negative for that antigen, as well. To make matters even more challenging, if the patient has developed anti-HLA antibodies (most commonly in previously pregnant females), then the donor should be HLA matched or at least HLA compatible with the patient’s antibodies. Finally, to complete the weirdness, it is essential to remember which modifications CAN and CAN’T be done to granulocytes. First, granulocytes CAN (and must) be irradiated to prevent Transfusion-associated Graft vs Host Disease (TA-GVHD). This is an extremely fresh product, full of highly active T-lymphocytes in addition to the granulocytes, and the recipient is immunocompromised, by definition. As a result, irradiation is essential to deactivate those donor T-lymphocytes and protect the patient (see image below). Second, granulocytes CAN’T be leukocyte-reduced. Every now and then, people will ask me about leukoreducing granulocytes for prevention of Cytomegalovirus (CMV) transmission. Rather than make fun of them, I usually just sit quietly while they work it out themselves (“OK, let’s see: Granulocytes are WBCs, which some people call ‘leukocytes,’ so if I leukocyte-reduce a unit of a product that is primarily composed of leukocytes, I would be left with…nothing! OHHHH!”). As a result of our inability to make a granulocyte product “CMV-safe” by leukoreduction, CMV-seronegative donors will be recruited to provide products for CMV-seronegative patients whenever possible. Note that it is totally fine to run granulocyte concentrate through a “standard” transfusion filter, just not a leukoreduction or microaggregate filter.


Irradiation is not only OK for granulocyte concentrate, it is essential! Leukocyte reduction, on the other hand, makes no sense for granulocytes, and shouldn’t be done.
5. Granulocytes very commonly cause reactions in recipients. Granulocytes are famous for causing transfusion reactions much more often than other components. The two issues that are seen most often are fever and chills (without hemolysis) and pulmonary reactions. I won’t discuss the self-explanatory fever and chills issue, but the pulmonary reactions are worth mentioning here. Considering that current thought suggests that neutrophils are the primary cell involved in the pathogenesis of Transfusion-related Acute Lung Injury (TRALI), it is not surprising that granulocyte transfusions are thought to cause pulmonary compromise quite often (5% in one study). Granulocytes LOVE the lungs, and they localize there very quickly (especially when the infection being treated is pneumonia). So, even if full-blown TRALI does not occur (which certainly CAN happen!), granulocyte transfusions are notorious for at least transient pulmonary compromise. Clinicians should be prepared to manage dypsnea and hypoxia during granulocyte transfusion. Note that the previously suggested “link” between more pulmonary reactions when granulocytes were given with the antifungal Amphotericin B seems to be disproven (though many still advise avoiding transfusing granulocytes within a few hours of administration of that medication). As mentioned in item 4, TA-GVHD is a risk for a fresh product, so irradiation is required. Further, HLA alloummunization can certainly occur as a result of granulocyte transfusion. Final thoughts: There is no question to me that granulocytes CAN, in some circumstances, appear to make a difference for patients. Unfortunately, there’s not a lot of objective proof that they are a predictably helpful treatment. However, given our current ability to collect granulocyte products with substantially higher yields than in years past, I expect that interest in this product will continue despite the lack of “proof” of its effectiveness.
I hope that this quick “5 Things to Know” has been useful for you!

-Joe Chaffin, MD, is the new Vice President and Chief Medical Officer for LifeStream, a Southern California blood center headquartered in San Bernardino, CA. He has a long history of innovative educational efforts and is most widely known as the founder and chief author of “The Blood Bank Guy” website (www.bbguy.org).