A critical task for clinical microbiologists is interpreting antimicrobial susceptibility testing (AST) results for microorganisms recovered from patient specimens. But what happens to that information after it is passed along to the clinical team, and how does it influence patient care at the bedside? In our previous blog, we discussed details concerning how breakpoints are established and identified them as an indispensable component of appropriate and effective antimicrobial prescribing. Here we will discuss the more practical application of breakpoints to guide patient care. To aid in the discussion, I’ve once again recruited two infectious diseases (ID) pharmacists from the UT Southwestern Medical Center to provide their valuable prospective on the use of AST results both at the bedside and to optimize antimicrobial stewardship initiatives.
Applying Breakpoints in Clinical Care
In the most simplistic terms, the reporting of isolate’s categorical susceptibility (susceptible, intermediate, and resistant) provides guidance concerning which antimicrobials will likely be effective to treat an infection.1 Many microbiology laboratories, however, report additional AST information including minimal inhibitory concentrations (MICs) and phenotypic information concerning resistance mechanisms (e.g. production of Extended Spectrum Beta-Lactamases (ESBLs) or inducible-clindamycin resistance)2. This more detailed information is often utilized by ID specialists to help guide and optimize beside management.
Unfortunately, several misapplications of AST data can be encountered in clinical care (Image 1). One common error is simply selecting the antimicrobial with the lowest MIC for treatment. This runs contrary to the idea that each antimicrobial has an individualized breakpoint for a given pathogen or pathogen group. Therefore, a low MIC for antimicrobial “A” may be at or near the breakpoint, but higher MIC for antimicrobial “B” may be a dilution or more lower than the breakpoint. Another common misconception is that if the isolate is reported susceptible to a given antimicrobial, that antimicrobial is the optimal choice for the patient, and will work 100% of the time! Very little in medicine is 100%, and this holds true for antimicrobials. Even if the optimal agent is selected based on AST data, there is no guarantee it will be effective in the patient. Many factors influence the effectiveness of antimicrobials including patient characteristics (e.g., immune system, kidney function), drug characteristics (i.e., pharmacokinetics [PK]/pharmacodynamics[PD]), and bug characteristics (e.g., underlying resistance present despite reporting as susceptible or potential for inducible resistance).2 This illustrates why clinicians may prefer to have all available information to further aid in optimization of antimicrobial therapy beyond the information provided by categorical reporting.
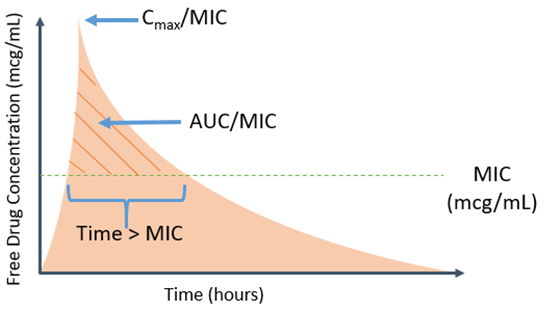
But why is an MIC needed – isn’t just knowing something is susceptible or resistant enough? In particular, more granular information such as the actual MIC for a given bug-drug combination may aid in more intricate decisions surrounding the choice of agent and dose. One argument for routine reporting of MIC values is the ability to better discern how near the breakpoint the particular MIC is for a given drug. The standard MIC reporting error is plus or minus one doubling dilution. When this is factored in, it may provide additional context as to how near or over the isolates phenotype is to assigned breakpoint.2 Clinicians may further couple this with their understanding of PK/PD to determine the optimal agent based on antimicrobial properties and microbial characteristics.
A counter argument is that the MIC values are prone to variation depending on testing modality and may not represent truly what occurs in actual humans (see Mouton JW et al. for more detailed discussion).3 As mentioned above, some argue that MIC reporting leads to misinterpretations of AST by a provider’s tendency to pick the lowest number, and thus prefer categorical reporting for simplification. Provider education and selective or cascade reporting are two strategies that may help clinicians select the optimal agent, but also have inherent limitations. All this taken into consideration, we feel the benefit of antimicrobial optimization supports reporting the MIC.
Applying Breakpoints at the Bedside
Breakpoints and MIC data are utilized at the bedside to avoid utilization of suboptimal agents due to issues with the drug and bug. For example, underlying resistance patterns may not be overtly apparent to clinicians. Depending on an institution’s reporting criteria, an E. coli isolate may be reported as susceptible to piperacillin/tazobactam despite being identified as an ESBL producer. In this instance, piperacillin/tazobactam may be suboptimal for certain infections based on available outcomes data, with some supposition that reported MIC values may be responsible for improper drug selection and treatment failures.4 Therefore, clinicians may favor alternative agents for infections despite documented susceptibility. This example also illustrates that certain resistance patterns (e.g., ceftriaxone resistance as surrogate for possible ESBL production) may help alert clinicians to underlying resistance, steering them clear of potential suboptimal agents reported as susceptible.
ID pharmacists often utilize MIC values to determine the optimal agent and dose for a given patient based on a working knowledge of breakpoint relationships and PK/PD. This may be taken a step further in the setting of an infection with a multidrug resistant organism which exhibit MICs in the intermediate or susceptible dose-dependent range. For certain drug-bug combinations with elevated MICs, it may be possible for the ID pharmacist to devise an optimized and personalized therapeutic regimen that will likely overcome the elevated MIC, either as monotherapy or utilizing potential synergistic combinations of different antibiotics.
The application of dose-dependent breakpoints, as seen with daptomycin for Enterococcus faecium, help illustrate the importance of understanding and applying dosing concepts in the context of established clinical breakpoints.1 This allows the clinician to not only optimize dosing, but also understand that an MIC at or near the breakpoint may warrant combination therapy in the setting of a serious infection. A lack of understanding of the relationship between breakpoints and dosing could potentially lead to an inadvertent assumption that lower FDA-approved doses would be reasonable despite the breakpoint being based on higher dosing strategies. These are just a small sampling of the clinical applications utilized on a daily basis to optimize a particular patient’s antimicrobial therapy.
A more global application of breakpoints occurs within antimicrobial stewardship programs. Antimicrobial stewardship focuses on the appropriate and optimal use of antimicrobials. To aid in this goal, AST reporting is key to developing clinical guidance on treating a variety of infections. Most notably, the optimization of empiric antimicrobial choices is aided by local or institutional susceptibility patterns. The cumulative antibiogram is pivotal to establishing optimal empiric antimicrobial choices across the spectrum of infectious diseases.5 Importantly, breakpoints are revised when new or additional clinical information becomes available, further illustrating the need to understand how a given institution implements and applies them. There is always the potential that from one year to the next susceptibilities may drop substantially due to implementation of new breakpoints, not necessarily an increased incidence of pathogens with a certain resistance pattern.6 In addition, the ability to adopt CLSI breakpoints often lags well beyond the release of the updated breakpoints, necessitating clinicians to be cognizant of changes and carefully assess categorical reporting as it may be discordant with the most up-to-date breakpoints.1,6 This topic will be the focus of our next blog.
More specific application of AST data can be made for either site (e.g., blood, urine, etc.), infection type (e.g., community-acquired pneumonia), or organism-specific MIC distributions. A recent trend in national guideline recommendations centers on ascertaining local susceptibility and pathogen distribution to provide optimal antimicrobial choices for certain infections (e.g., pneumonia).7 In addition, focused review of individual pathogen MIC distributions provide another level of detail that helps better define upfront institutional antimicrobial dosing strategies to optimize PK/PD parameters. In conclusion, the reporting of AST data remains a cornerstone for the effective management of infections. Information collected by laboratory technicians provides invaluable information to clinicians. This information helps optimize the selection and dosing of antimicrobial agents at the bedside, which in turn improves the clinical management of patients and infection-related outcomes.

References
- Clinical and Laboratory Standards Institute. Development of In Vitro Susceptibility Testing Criteria and Quality Control Parameters, 5th Edition (M23).
- Giuliano C, Patel CR, and Kale-Pradhan PB. A Guide to Bacterial Culture Identification and Results Interpretation. P&T. 2019 April 44(4):192-200.
- Mouton JW, Muller AE, Canton R, et al. MIC-based dose adjustment: facts and fables. J Antimicrob Chemother 2018; 73:567-68.
- Pogue JM and Heil EL. Laces out Dan! The role of tazobactam based combinations for invasive ESBL infections in a post-MERINO world. E Expert Opin Pharmacother.2019 Dec;20(17):2053-57.
- Clinical and Laboratory Standards Institute. Analysis and Presentation of Cumulative Antimicrobial Susceptibility Test Data; Approved Guideline, 4th Edition (M39-A4).
- Humphries RM, Abbott A, Hindler JA. Understanding and Addressing CLSI Breakpoint Revisions: a Primer for Clinical Laboratories. J Clin Microbiol. 2019 May 24;57(6):e00203-19.
- Khalil AC, Metersky ML, Klompas M, et al. Management of Adults with Hospital-acquired and Ventilator-associated Pneumonia: 2016 Clinical Practice Guidelines by the Infectious Diseases Society of America and the American Thoracic Society. Clin Infect Dis. 2016 Sep 1;63(5):e61-e111.

-James Sanders, PharmD, PhD, is an infectious diseases pharmacy specialist and assistant professor at the University of Texas Southwestern Medical Center in Dallas, Texas. His academic and research interests are focused on multi-drug resistant Gram-negative bacteria, surgical site infections and HIV pharmacotherapy.

-Marguerite Monogue, PharmD is an infectious diseases pharmacy specialist and assistant professor at the University of Texas Southwestern Medical Center in Dallas, Texas. She is interested in antimicrobial pharmacokinetics/pharmacodynamics and multi-drug resistant Gram-negative bacteria.

-Andrew Clark, PhD, D(ABMM) is an Assistant Professor at UT Southwestern Medical Center in the Department of Pathology, and Associate Director of the Clements University Hospital microbiology laboratory. He completed a CPEP-accredited postdoctoral fellowship in Medical and Public Health Microbiology at National Institutes of Health, and is interested in antimicrobial susceptibility and anaerobe pathophysiology.