Working bacteriology benches in the clinical microbiology laboratory often comes with its fair share of questions about the susceptibility of patient isolates. In training, we are taught about breakpoints – clinically essential values which determine if an organism is susceptible, resistant, or somewhere in-between for a given drug. These values are readily accessible to us in guidance documents (e.g., CLSI M100, FDA website), and are programmed into instruments to allow for automated interpretation. But, have you ever wondered how these values are derived? Raw microbiological and pharmacological data, patient outcomes, and regulatory considerations must be examined through multiple lenses, and by many different entities, before these values ever make it to the printed page and used clinically. Here, we will highlight some of the “moving parts” of breakpoint determination an effort to demystify this process and gain a better understanding and appreciation of the clinical application of these life-saving measurements. To help, I’ve recruited two of our outstanding infectious disease pharmacists from UT Southwestern Medical Center to enhance this discussion. It’s my hope we will all learn something in the process!
Breakpoint Breakdown – The ABC’s of MICs and ECVs, plus Pharmacology 101!
A breakpoint represents a defined antibiotic concentration or zone of inhibition diameter that serves as a gatekeeper for antimicrobial use. This value categorizes organisms as susceptible, susceptible-dose dependent, intermediate, resistant, or nonsusceptible to various antimicrobials. As such, is an indispensable component of appropriate and effective antimicrobial prescribing.1 Breakpoints are set through a rigorous examination of data by various national and international organizations which we will discuss in a later post. Determining the optimal value at which a breakpoint should be set is multifactorial, requiring a multidisciplinary approach to incorporate data from bench and bedside. Now, if that introduction sounded daunting, don’t panic! We’re going start at the beginning with basic biological measurements of susceptibility that are needed to begin to establish a breakpoint. For those not currently working in microbiology (or if it’s been a while), a basic understanding of susceptibility testing mechanisms is necessary and we will briefly review here. Bacteria will be the focus for this discussion, but many of these concepts are also broadly applicable to fungi as well.
Determination of susceptibility to an antibiotic can be evaluated by examining the response of a bacterial isolate to antimicrobial exposure. In the laboratory, this is usually achieved through dilution, whereby an isolate may grow at some drug concentrations, and growth is inhibited at others. Dilution of the antimicrobial can come either through directly applying the antibiotic at defined concentrations uniformly to growth media (i.e. broth dilution, agar dilution), or utilizing a diffusion gradient through media when an antibiotic is applied at a single source (i.e. disk diffusion) (Image 1). Application of defined antibiotic concentrations to the growth media allows for a minimal inhibitory concentration (MIC) to be determined, while a diffusion gradient allows for a zone of inhibition to be measured (ZOI). An MIC is the lowest concentration of antimicrobial that inhibits organism growth of an isolate. This value is unique to the isolate that is being tested. However, when establishing a broad measurement which will encompass all isolates of that species (or group of species) such as a breakpoint, it’s easy to imagine that significantly more data is needed.

Thus, a first step in setting an optimal breakpoint begins with an antimicrobial’s in vitro activity against an organism. A descriptive summary of the MIC range across a given species helps define the MIC distribution. This analysis usually includes MICs from hundreds of tested isolates! This MIC distribution helps to define epidemiologic cutoff values (ECV or ECOFF). Like a breakpoint, these values separate the MIC distribution into bacterial populations that are either wild-type, and those with resistance. The wild-type MIC distribution aims to exclude outlier MICs that may represent organisms with acquired resistance (either through mutation or acquisition of resistance determinants) reflected by elevated MICs. An isolate in the population with an MIC above the ECV is likely to have acquired resistance, whereas an isolate with an MIC lower than the ECV likely originates from the wild-type distribution and lacks mechanisms of acquired resistance or reduced susceptibility.2 So great, we have an experimental MIC value which separates wild-type organisms from ones that have acquired resistance, why not just stop there and call it a breakpoint? The answer is the ECV does not account for host responses, clinical outcomes, site of infection, pharmacokinetics/pharmacodynamics, dosing, and a number of other important variables which go into establishing a clinical breakpoint. Thus, using ECVs for clinical decision making is challenging.
Now that we have considered some aspects of the microbiological side of the equation, let’s switch gears and look at the host and other factors not addressed by an ECV. Pharmacokinetic (PK) and pharmacodynamic (PD) parameters are key to assessing the clinical applicability of a breakpoint. PK parameters represent how the body handles the antimicrobial, including absorption, distribution, metabolism and elimination, whereas PD parameters represent the effect between the drug and bug. Taken together, PK/PD parameters represent the relationship between drug concentration (PK) and antimicrobial effect (PD) over time. Different antimicrobials have distinct PK and PD characteristics, thus several PK/PD indices are utilized to determine optimal target concentrations or exposures that improve antibacterial efficacy. Common PK/PD indices are the percent of time the free drug concentration remains above an organism’s MIC (fT > MIC), the ratio of free max drug concentration (or peak) to MIC (fCmax/MIC), and the ratio of free drug exposure (area under the curve, AUC) over a 24-hour period to MIC (fAUC/MIC) (Image 2).3
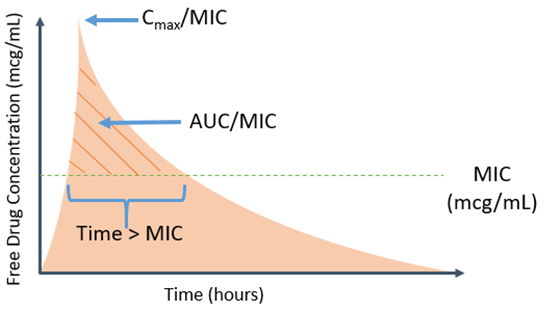
For example, β-lactam antimicrobials require 40-60% fT > MIC for maximum antibacterial efficacy; however, the exact fT > MIC (or other exposure) required for optimal efficacy varies between different β-lactam antimicrobials and bacterial species. Importantly, the desired antimicrobial exposure should be obtainable based on the established breakpoint, meaning the β-lactam of interest should achieve 40-60% fT > breakpoint. Various strategies are employed to optimize PK/PD parameters relative to a given breakpoint (more on that to come in Part 2 of this series). These target exposures are calculated from data for each bug-drug combination.
Of note, the antimicrobial exposure in relation to MIC is often based on antimicrobial blood concentrations. However, this is obviously not always the site of infection. The same bug-drug combination can have multiple breakpoints specific to infection site. For example, central nervous system (CNS) infections may have lower established breakpoints compared with non-CNS infections. The use of a lower breakpoint improves PK/PD target attainment in areas where there may be low drug concentrations, such as the CNS. For CNS-specific breakpoints, only organisms with MICs within the lower range of the MIC distribution will be deemed susceptible. With a lower MIC, the PK/PD exposure may be more obtainable given the potential poor drug penetration to the site of infection.
Outcomes from clinical data are often deemed the most important factor used to determine breakpoints. Treatment successes or failures of an antimicrobial against a specific MIC may provide validity to an established breakpoint or support revision. For example, the piperacillin-tazobactam breakpoint for Pseudomonas aeruginosa, was lowered from 64/4 mcg/mL to 16/4 mcg/mL due to an increase in mortality observed in patients who had organisms with MICs 32 or 64 mcg/mL.4 Unfortunately, clinical outcome data is often influenced by other confounders beyond antimicrobial therapy and the organism’s MIC, such as source control and other therapeutic interventions. Furthermore, clinical data for “resistant” organisms may not be available, limiting the assessment of the antimicrobial against organisms with high MICs.
Finally, it is important to remember that breakpoints are not set in stone and change regularly as more data become available. Common reasons for breakpoint revisions include: 1) new PK/PD data suggesting the breakpoint is too low or high based on antimicrobial exposure, 2) identification of novel resistance mechanisms, and 3) new clinical data to suggest poor correlation of clinical response with the established breakpoint. Furthermore, microbiological methods may become more accurate and ultimately affect quantification of the MIC.5
In summary, establishing a breakpoint is not a straightforward process and requires an aggregate of information. We are really only scratching the surface of the very complex process here, but hope that this sheds some light on where these important values originate. Various types of data; microbiological, pharmacological, in vitro, in vivo, they all create the story. However, a complete picture is often not available at the time of breakpoint creation, resulting in the need to constantly review and update breakpoints as more data becomes available.
Next post we will discuss how breakpoints are used at the bedside.
References
- Clinical and Laboratory Standards Institute. Development of In Vitro Susceptibility Testing Criteria and Quality Control Parameters, 5th Edition (M23).
- Turnidge J, Paterson DL. Setting and revising antibacterial susceptibility breakpoints. Clin Microbiol Rev. 2007 Jul;20(3):391-408.
- Craig WA. Pharmacokinetic/pharmacodynamic parameters: rationale for antibacterial dosing of mice and men. Clin Infect Dis. 1998 Jan;26(1):1-10; quiz 11-2. doi: 10.1086/516284.
- Tam V, et al. Outcomes of bacteremia due to Pseudomonas aeruginosa with reduced susceptibility to piperacillin-tazobactam: implications on the appropriateness of the resistance breakpoint. Clin Infect Dis. 2008 Mar 15;46(6):862-7.
- Humphries RM, Abbott A, Hindler JA. Understanding and Addressing CLSI Breakpoint Revisions: a Primer for Clinical Laboratories. J Clin Microbiol. 2019 May 24;57(6):e00203-19.

-Marguerite Monogue, PharmD is an infectious diseases pharmacy specialist and assistant professor at the University of Texas Southwestern Medical Center in Dallas, Texas. She is interested in antimicrobial pharmacokinetics/pharmacodynamics and multi-drug resistant Gram-negative bacteria.

-James Sanders, PharmD, PhD, is an infectious diseases pharmacy specialist and assistant professor at the University of Texas Southwestern Medical Center in Dallas, Texas. His academic and research interests are focused on multi-drug resistant Gram-negative bacteria, surgical site infections and HIV pharmacotherapy.

-Andrew Clark, PhD, D(ABMM) is an Assistant Professor at UT Southwestern Medical Center in the Department of Pathology, and Associate Director of the Clements University Hospital microbiology laboratory. He completed a CPEP-accredited postdoctoral fellowship in Medical and Public Health Microbiology at National Institutes of Health, and is interested in antimicrobial susceptibility and anaerobe pathophysiology.
