Sounds like a good title for a sci-fi novel, right? What is the big deal about Next Generation Sequencing (NGS)? Otherwise known as massively parallel sequencing or high throughput sequencing, NGS has become a technique used by many molecular labs to interrogate multiple areas of the genome in a short amount of time with high confidence in the results. Throughout the next few blogs, we’ll discuss why NGS has become the next big thing in the world of molecular. We’ll go through the steps of setting up the specimens to prepare them to be sequenced (library preparation), what types of platforms are available and what technologies they use to sequence. Lastly, we’ll go through some of the challenges with this type of technology.
Let’s start with a review of dideoxy sequencing, otherwise known as Sanger sequencing, which has been the gold standard since its inception in 1977. A typical setup in our lab for this assay begins with a standard PCR to amplify a region of the genome that we are interested in, say PIK3CA exon 21, specifically amino acid 1047, a histidine (CAT). The setup would include primers complementary to an area around exon 21, a 10x buffer, MgCl2, a deoxynucleotide mix (dNTP’s), and Taq polymerase. After amplification, the resulting products would be purified with exonuclease and shrimp alkaline phosphatase (SAP). Next, another PCR would be set up using the purified products as the sample and using a similar mix as in the original amp, but with the addition of a low concentration of fluorescently labeled dideoxynucleotides. These bases have no -OH group, so when they are incorporated into the product, amplification ceases on that strand. Because they are present in a lower concentration, the incorporation of these is random and will occur at each base in the strand eventually. The resulting products are then run and analyzed on a capillary electrophoresis instrument that will detect the fluorescent label on the dideoxynucleotides at the end of each fragment. Shown below is an example of the output of the data:

The bases will be shown as peaks as they are read across the laser. The base in question in the middle of the picture is, in a “normal” sequence, an adenine (A), as seen in green. In this case, there is also a thymine (T) detected at that same location, as seen in red. This indicates that some of the DNA in this tumor sample has mutated from an A to a T at this location. This causes a change from a histidine amino acid to a leucine (p.His1047Leu) and is a common mutation in colorectal cancers.
So all of this looks great, right? Why do we need to have another method since we have been using this one for so long and it works so well? There are a few reasons:
- The sensitivity of dideoxy sequencing is only about 20%. This means lower level mutations could be missed. The sensitivity of NGS can get down to 5% or even lower in some instances.
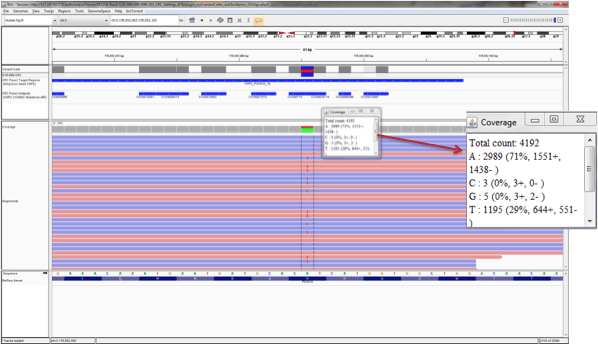
- The above picture shows the sequencing in the forward direction as well as the reverse direction. This area then has 2x coverage – we can see the mutation in both reads. If we could get a higher coverage of this area and be able to sequence it multiple times and see that data, we could feel more confident that this mutation is real. In our lab, we require each area has 500x coverage so that we feel sure that we have not missed anything. The picture below displays the same sequenced area as in the dideoxy sequencing above. This a typical readout from an NGS assay and, as you can see, this base has a total of 4192 reads, so it has been sequenced over four thousand times. In 1195 of those reads, a T was detected, not an A. We can feel very confident in these results due to how many times the area was covered.
- The steps above detailed only amplifying this one area, but with colorectal cancer specimens, we want to know the status of the KRAS, BRAF, NRAS, and HRAS genes as well as other exons in PIK3CA Using the dideoxy sequencing method is a lot of time and effort. NGS can cover these areas in these five genes as well as multiple other areas (our assay looks at 207 areas total) all in the same workflow
Join me for the next installment to discover the first steps in NGS workflow!

-Sharleen Rapp, BS, MB (ASCP)CM is a Molecular Diagnostics Coordinator in the Molecular Diagnostics Laboratory at Nebraska Medicine.

