Case History
A 48-year-old female presents with a one-month history of left upper quadrant pain. Laboratory investigation reveals pancytopenia. Radiology work-up demonstrates splenomegaly. CT scan confirms splenomegaly at 22 cm. There is no lymphadenopathy appreciated in the abdomen. A bone marrow biopsy is performed.
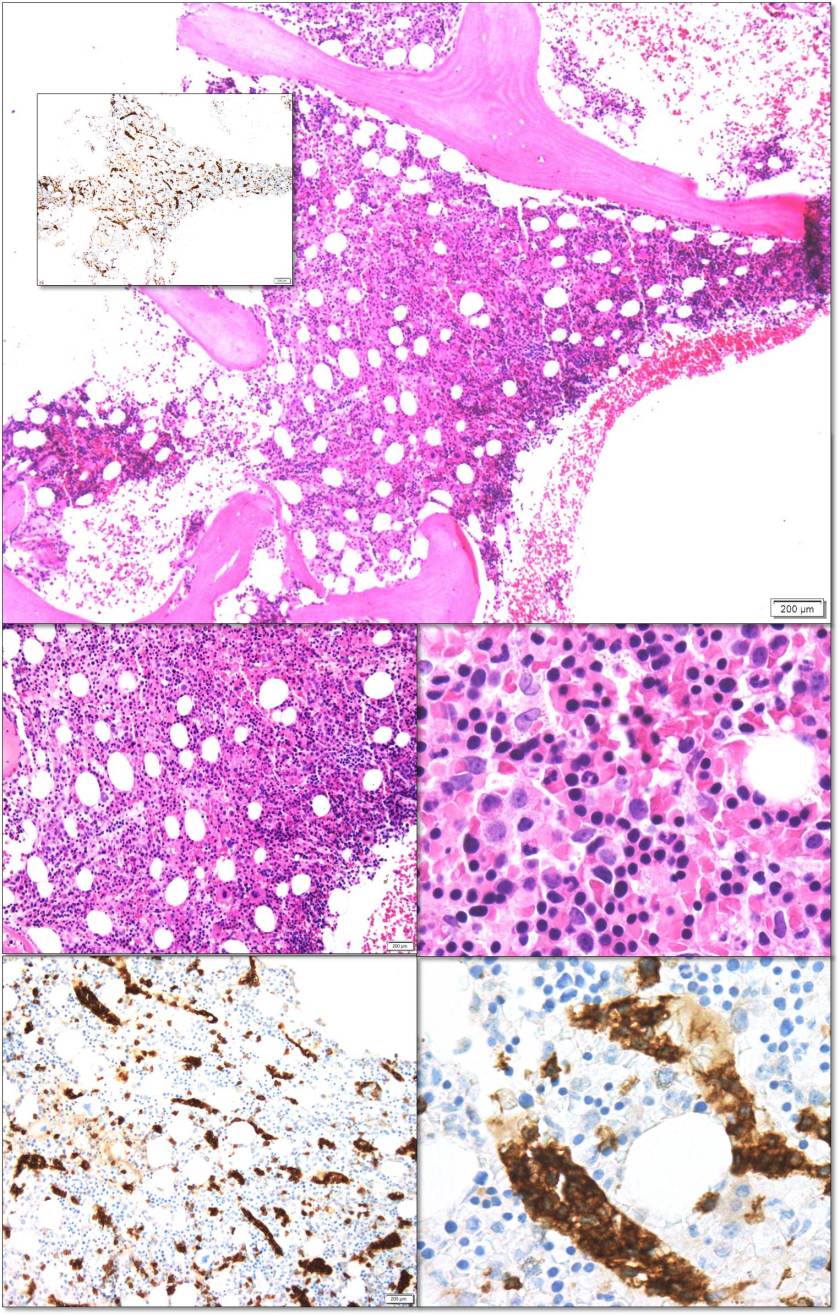
Bone Marrow Findings
The bone marrow core biopsy reveals a normocellular marrow space (approximately 50% cellular marrow) with progressive trilineage hematopoiesis. Clusters of small, slightly irregular, mature-appearing lymphocytes are seen within the sinusoids. The marrow aspirate smears reveal mild erythroid hyperplasia without morphologic evidence of dysplasia. There is no increase in blasts. Lymphocytes comprise 18% of a 500-cell differential count on the marrow aspirate smears.
The sinusoidally distributed lymphocytes demonstrate immunopositivity (flow and/or IHC) for CD2, CD3, CD7, CD16, CD56, and γδ. These neoplastic lymphocytes are negative for granzyme B, CD4, CD5, CD8, CD57, and αβ. PCR for T-cell receptor clonality was positive. Cytogenetics revealed a normal female karyotype. FISH for 5p/5q and 7p11/7q31 was normal.
Diagnosis
Taken together, the patient’s clinical presentation along with the presence of an abnormal gamma-delta population of T cells in a sinusoidal distribution with PCR evidence of T-cell clonality is diagnostic of a T-cell lymphoma. The pattern of distribution, granzyme B negativity, lack of concurrent adenopathy favor a diagnosis of Hepatosplenic T-cell lymphoma.
Discussion
Hepatosplenic T-cell lymphoma (HSTCL) is an uncommon entity that represents <1% of all non-Hodgkin lymphomas and 1%-2% of all T/natural killer cell lymphomas. It most commonly affects young adult men, with a median age of 35 years. This high-grade malignancy is most often characterized by γδ T-cells. The most consistent symptoms among patients are fever, splenomegaly, hepatomegaly, bone marrow involvement, peripheral blood cytopenia, and less commonly, adenopathy. Hepatosplenic T-cell lymphoma has a poor prognosis with median survival rates varying from a few months to 16 months in different studies.
Immune suppression (such as solid- organ transplant, or immune dysregulation secondary to malignancy or infection) is thought to play a role in the lymphomagenesis in around 20% of cases. Inflammatory bowel disease and the use of immunosuppressive agents (e.g., antitumor necrosis- α agents) and antimetabolite therapy (e.g., 6TG, 6MP) had also been associated with development of HSTCL.
HSTCL initially infiltrates the cords and sinusoids of the splenic red pulp. The white pulp is often atrophic or absent. Eventually, the neoplastic T cells diffusely replace the spleen. The lymphoma cells often involve the liver and bone marrow sinusoids. At the time of diagnosis, the bone marrow is almost always involved and commonly hypercellular. The neoplastic cells are mostly intermediate in size, with pale agranular cytoplasm and round nuclei with condensed chromatin and inconspicuous nucleoli.
Cytogenetic studies in HSTCL most commonly show isochromosome 7q and trisomy 8. Molecular analysis of HSTCL characteristically shows expression of a γδ T-cell type and flow cytometric analysis typically reveals a CD2+, CD3+, CD7+/−, CD4−, CD5−, and CD8− phenotype with positivity for natural killer cell-associated markers CD11b, CD16, and CD56.
Activating mutations in PI3KCD and STAT signaling genes have also been described in HSTCL, providing potential molecular target therapies for this aggressive lymphoma.
The differential diagnosis of HSTCL includes other types of T-cell lymphoma and leukemia, and non-neoplastic such as immune thrombocytopenia or acute hepatitis. In most instances, the distinctive presentation of spleen, liver and bone marrow involvement, the immunophenotype and T-cell monoclonality distinguishes HSTCL from other entities.
The outcomes of the patients using standard chemotherapy regimens are dismal, and allogeneic SCT appears to be a reasonable approach to achieve the best possible patient outcome.
References
- Yabe M, Miranda RN, Medeiros LJ. Hepatosplenic T-cell Lymphoma: a review of clinicopathologic features, pathogenesis, and prognostic factors. Hum Pathol. 2018 Apr; 74:5-16.
- McThenia SS, Rawwas J, Oliveira JL, Khan SP, Rodriguez V. Hepatosplenic γδ T-cell lymphoma of two adolescents: Case report and retrospective literature review in children, adolescents, and young adults. Pediatr Transplant. 2018 Aug;22(5): e13213.

-Levent Trabzonlu, MD is a postdoctoral researcher in the department of pathology at Johns Hopkins University in Baltimore, MD. Follow Dr. Trabzonlu on twitter @aflevent

-Kamran M. Mirza, MD PhD is an Assistant Professor of Pathology and Medical Director of Molecular Pathology at Loyola University Medical Center. He was a top 5 honoree in ASCP’s Forty Under 40 2017. Follow Dr. Mirza on twitter @kmirza.
