Case History
A 20 year old woman with cystic fibrosis was routinely screened with bacterial sputum cultures. The patient reported feeling well and was compliant with her treatment regimen. She had no history of Nocardia colonization in the past and her last hospitalization was four years prior due to a pulmonary exacerbation in which cultures grew Stenotrophomonas and Pseudomonas.
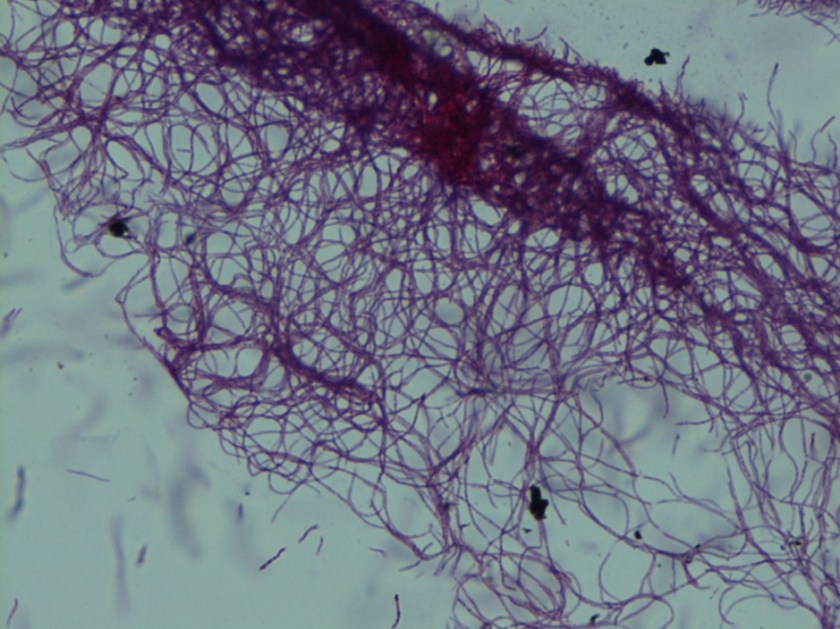
Image 1: Modified acid fast stain showing two unique traits of Nocardia: long, delicate rods and weak acid fastness.

Image 2: Nocardia colonies on buffered charcoal yeast extract (BCYE) agar.
Discussion
Nocardia are slow growing, aerobic gram negative rods. Nocardia are unique for being weakly acid fast and displaying aerial hyphae. They are ubiquitous organisms that can cause a variety of infections in immunocompromised individuals. Most often in the United States, nocardiosis causes a lung infection. If left untreated, it can spread to the brain or spinal cord, where up to 44% die [1].
The above patient showed no signs of an infection, making the possibility of colonization by Nocardia more likely. Cystic fibrosis patients can be colonized by Nocardia and the clinical approach is to treat regardless of the patient’s overall health. However, there is a lack of reporting on whether Nocardia is the cause of an infection when it happens. Host and pathogen interactions are also not well known. Thorn et al. showed that treating cystic fibrosis patients that are colonized with Nocardia with oral antibiotics did not affect their clinical outcome [2]. More studies are needed to be done to see if antibiotics are warranted in circumstances like the above patient.
References:
- Pulmonary nocardiosis in cystic fibrosis. Thorn, Shannon T. et al.. Journal of Cystic Fibrosis, Volume 8 , Issue 5 , 316 – 320
-Angela Theiss is a pathology resident at the University of Vermont Medical Center.

-Christi Wojewoda, MD, is the Director of Clinical Microbiology at the University of Vermont Medical Center and an Associate Professor at the University of Vermont.
