This month, I’m switching gears to another interest of mine: Molecular Pathology. I am currently in fellowship for Molecular Genetic Pathology which exposes me to unique, thought-provoking cases.
Advances in genomic sequencing has allowed multiple genes to be analyzed in a single laboratory test. These so-called gene panels have increased diagnostic yield when compared to serial gene sequencing in syndromic and non-syndromic diseases with multiple genetic etiologies. However, interpretation of genetic information is complicated and evolving. This has led to wide variation in how results are reported. A genetic test result can either be positive (pathogenic or likely pathogenic), negative (benign or likely benign) or uncertain (variant of uncertain significance- VUS). A VUS may just be part of what makes each individual unique and doesn’t have enough evidence present to say that it is pathogenic or benign. Many results come back like this and can be frustrating for patients to hear and for genetic counselors and clinicians to explain.
Initial approaches to exclude benign variants through sequencing 100 “normal people” to determine the frequency of common variants in the population was fraught with bias. The “normal population” initially was constructed mostly of individuals with white European descent. Not surprisingly, lack of genetic diversity in control populations lead to errors in interpretation.
Fortunately, there are now several publicly available databases that exist to help determine whether gene variants are damaging. The first important piece comes from population sequencing efforts. These projects performed whole exome sequencing of hundreds or thousands of individuals to find variants that might be rarely expressed in a more genetically diverse population. If a variant occurs in a normal health population at a frequency >1%, then it likely doesn’t cause a severe congenital disease that would in turn prevent that genetic variant from being passed on.
The Exome Association Consortium (ExAC)1, which has been rolled into the larger gnomAD (genome aggregation database) database now contains sequencing information on 120,000 individuals (Figure 1). The smaller ESP (Exome Sequencing Project) was a project by the NHLBI division of NIH and sequenced several patients with different cardiovascular and pulmonary diseases.

While there is ethnic diversity present in this database, the 1000 genomes project2 furthered efforts by searching all over the world to get genetic information from around 100 ethnically and geographically distinct sub-populations (Figure 2).
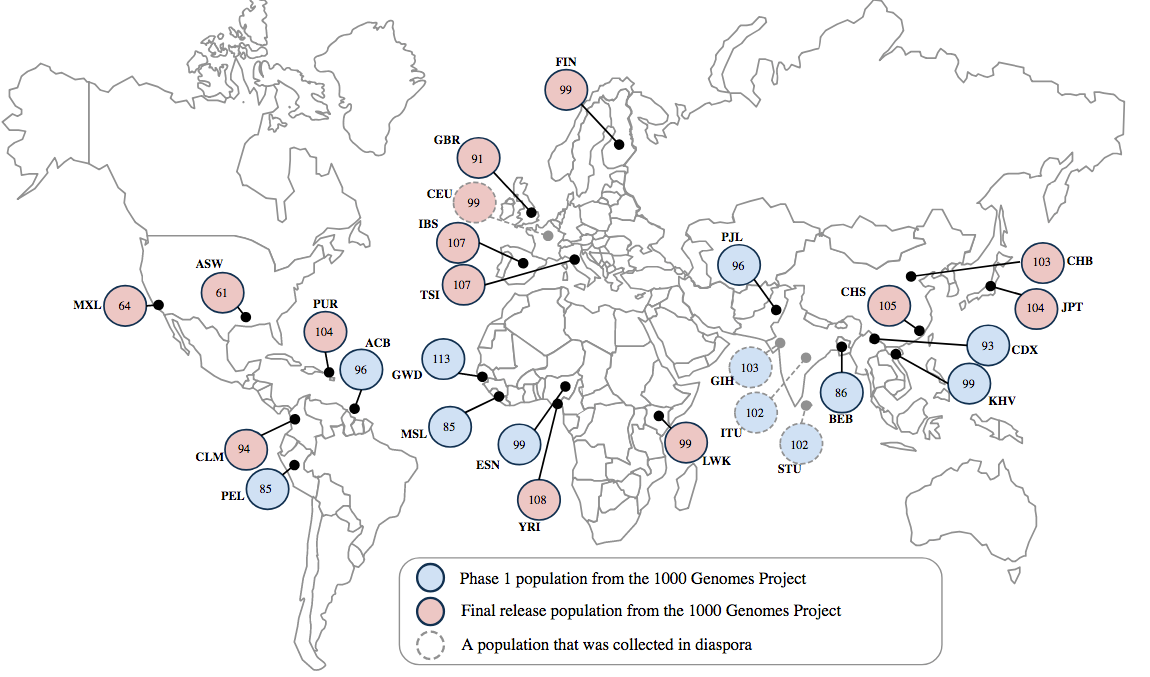
With use of these databases, we can effectively rule out rare polymorphisms as benign when they are expressed in several healthy individuals and especially when expressed in the homozygous state in a healthy individual. Before, it was common for a person of an ethnic minority to have different variants compared to predominantly European cohorts. In many cases, this led to uncertain test results.
One way to deal with these VUSs is for a lab to periodically review their test results in light of new knowledge. Although the CAP has a checklist3 item that requires a lab to have a policy about reassessing variants and actions taken. However, this item doesn’t require a lab to communicate the results with a physician and doesn’t specify how often to reanalyze variants. Before last year, there weren’t even any studies that indicated how often variant reanalysis should occur. Variant reanalysis had only been studied in a limited context of whole exome sequencing for rare diseases to improve the diagnostic yield4. However, this did not address the issue of frequent VUSs to determine how often they were downgraded to benign or upgraded to pathogenic.
One example of how reclassification can occur is illustrated in the case of a young African American boy who had epilepsy and received a genomic test that covered a panel of genes known to be involved in epilepsy in 2014. Two heterozygous VUS were reported back for EFHC1 (EFHC1 c.229C>A p. P77T and EFHC1 c.662G>A p. R221H), which causes an autosomal dominant epilepsy syndrome when one allele is damaged. However, this variant could later be reclassified as benign by looking at population databases. The ExAC database showed an allele frequency of 2.5% in African Americans and the 1000 Genomes database showed an 8.8% frequency in the GWD subpopulation (Gambian Western Divisions).
This case demonstrates the importance of reanalyzing genetic test results as medical knowledge continues to evolve. Recently studies looking at reclassification rates of epilepsy5 and inherited cancer syndromes6 have been published in JAMA journals and demonstrate that reclassification of variants is common. It is thus important for laboratories to periodically review previously reported variants to provide optimal quality results and patient care. I will elaborate on this further in the next blog post.
References:
- Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285-291.
- The 1000 Genomes Project Consortium, Auton A, Brooks LD, et al. A global reference for human genetic variation. Nature. 2015;526:68-74.
- Sequence Variants – Interpretation and Reporting, MOL.36155. 2015 College of American Pathologists (CAP) Laboratory Accreditation Program Checklist.
- Costain G, Jobling R, Walker S. Periodic reanalysis of whole-genome sequencing data enhances the diagnostic advantage over standard clinical genetic testing. Eur J Hu Gen. 2018.
- SoRelle JA, Thodeson DM, Arnold S, Gotway G, Park JY. Clinical Utility of Reinterpreting Previously Reported Genomic Epilepsy Test Results for Pediatric Patients. JAMA Pediatr. 2018 Nov 5:e182302.
- Mersch J, Brown N, Pirzadeh-Miller, Mundt E, Cox HC, Brown K, Aston M, Esterling L, Manley S, Ross T. Prevalence of Variant Reclassification Following Hereditary Cancer Genetic Testing. JAMA. 2018 Sep 25;320(12):1266-1274.

-Jeff SoRelle, MD is a Molecular Genetic Pathology fellow at the University of Texas Southwestern Medical Center in Dallas, TX. His clinical research interests include understanding how the lab intersects with transgender healthcare and advancing quality in molecular diagnostics.
This work was produced with the guidance and support of:

Dr. Jason Park, MD, PhD, Associate Professor of Pathology, UT Southwestern Medical Center

Dr. Drew Thodeson, MD, Child Neurologist and Pediatric Epileptologist
