Von willebrand disease is the most common inherited bleeding disorder. Partial lack of VWF (von Willebrand factor) causes mild or moderate bleeding tendency. Patients typically present with menorrhagia, bruising, bleeding from gums or after surgery. It is typically autosomal dominant with variable penetrance. Laboratory investigation reveals defective platelet adherence (PFA-100) or long bleeding time, subnormal levels of von Willebrand antigen and factor VIII in plasma and low ristocetin cofactor activity or VWF activity.
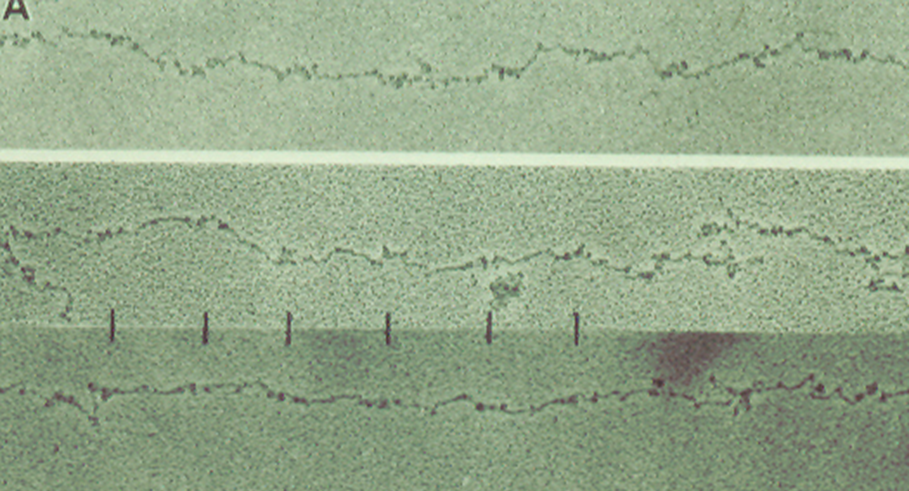
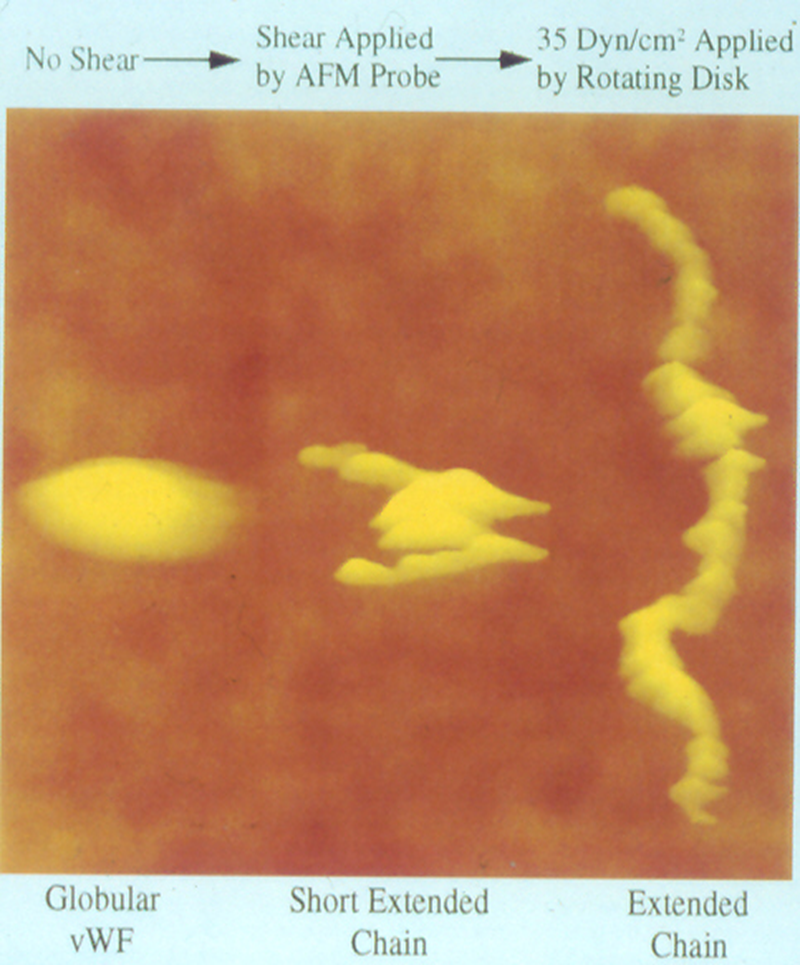
Blood, 1996; volume 88
Types of von Willebrand disease
- Type 3: Patient has severe deficiency of vWF. Antigen, activity and factor VIII levels are all < 10%. Clinically patients have a hemophilia-like phenotype. It is inherited recessively.
- Type 2: Patients have a qualitative defect (missense mutation) of the vWF. There are several different types. Usually there is a disproportionate decrease in vWF activity vs antigen.
- Type 1: vWF antigen and activity are reduced proportionately. vWF levels range from < 20% to ~50%. Only 65% of cases are associated with VWF gene mutations. It has an autosomal dominant inheritance pattern with variable penetrance (affected by blood type, other factors).Defects in VWF processing, storage or secretion may account for cases lacking VWF gene mutation. Some cases are associated with accelerated VWF clearance.
Type-2 vWD
- 2A: Deficiency of intermediate & large multimers due to either defective assembly (mutation in either of two domains involved in multimer formation), or increased susceptibility to proteolysis (mutation in domain cleaved by ADAMTS-13)
- 2B: Largest multimers are missing. This is due to gain of function mutation in platelet Gp Ib binding domain of vWF.Largest multimers bind spontaneously to platelets and are cleared from blood. Rarely, a mutation in Gp Ib may have the same effect (“platelet-type” vWD).
- 2M: Normal multimer pattern. There is loss of function mutation in GP Ib binding domain
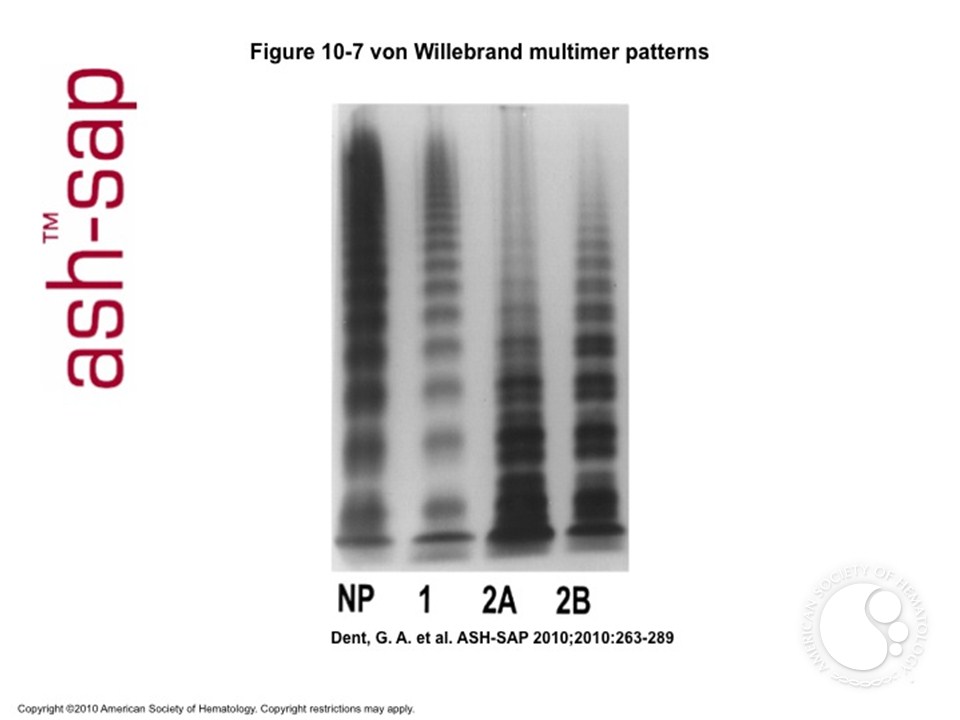
Laboratory analysis

Von Willebrand factor activity: Measures binding of patient VWF to latex beads coated with monoclonal Ab to GPIb binding site; sensitive to multimer size and platelet-binding ability
Platelet function screen (PFA): Measures time necessary for platelet plug to form in collagen coated tube under high shear conditions in the presence of ADP or epinephrine
Desmopressin (DDAVP) in VWD
DDAVP releases vWF from endothelial cells and can be given IV or intranasally ( 0.3 mcg/kg IV, or 150 mcg per nostril ). It typically causes 2-4 fold increase in blood levels of vWF (in type 1 vWD), with half-life of 8+ hours. Response to DDAVP varies considerably. Administration of a trial dose si necessary to ensure a given patient responds adequately.

Indications for clotting factor concentrate administration in vWD include
- Type 2 or 3 vWD with active bleeding, surgery or other invasive procedures or
- Type 1 vWD with inadequate response to DDAVP.
Acquired von Willebrand Disease
This can happen in association with either monoclonal gammopathy ( vWF neutralized by paraprotein) , autoimmune disorders (autoantibody to vWF), myeloproliferative disorder (large multimers absorbed onto neoplastic cells), cardiovascular diseases (AS, VSD, etc, high shear stress causes unfolding/proteolysis of large multimers), or hypothyroidism (decreased release of vWF from endothelial cells). Treatment varies depending on the cause/mechanism in each case.


-Neerja Vajpayee, MD, is an Associate Professor of Pathology at the SUNY Upstate Medical University, Syracuse, NY. She enjoys teaching hematology to residents, fellows and laboratory technologists.
